[0001] COMPOSITIONS AND METHODS FOR PREVENTING AND TREATING CARDIAC ISCHEMIA/REPERFUSION INJURY
[0002]FIELD OF THE INVENTION
[0003] The invention relates to compositions and methods of preventing and treating cardiac ischemia/reperfusion injury.
[0004]BACKGROUND OF THE INVENTION
[0005] A heart attack is usually caused by blockage of an artery. The heart muscle beyond the block is then deprived of oxygen and essential nutrients. This ischemia (literally "lack of blood flow") leads to many damaging changes, including calcium buildup in cells, high levels of reactive oxygen species ("ROS"), buildup of waste products such as lactic acid, and general energy depletion. These events may lead to cell death either by necrosis (i.e., directly from injury to the cells) or by apoptosis (i.e., from an energy dependent cell suicide process) and the formation of an infarct— a region of dead tissue. This process may be partially blocked by appropriate therapy. Re-establishment of blood flow (reperfusion) and re-oxygenation of the affected area is critical to limit irreversible damage. However, reperfusion also brings potentially damaging consequences.
[0006] Ischemia/reperfusion (I/R) injury refers to damage to tissue caused when blood supply returns to the tissue after a period of ischemia. The absence of oxygen and nutrients from blood creates a condition in which the restoration of circulation results in inflammation and oxidative damage through the induction of oxidative stress rather than restoration of normal function. Cardiac I/R is characterized by arrhythmias, cardiomyocyte damage, inflammation, and, at the cellular level, disturbance of Ca2+ and redox homeostasis.
[0007] Elevated plasma levels of tumor necrosis factor a (TNF-a) have been reported in cardiac ischemia/reperfusion injury, myocardial infarction and in congestive heart failure. TNF-a induces pleiotropic effects that are mediated through activation of two distinct receptors: TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2). Most of the deleterious effects attributed to TNF-a, including left ventricular remodeling, are mediated by TNFR1 signaling. TNF-a also has long-term beneficial effects due to the induction of cytoprotective genes involved in cellular growth, survival and proliferation, in response to pressure and volume overload. Thus, release of TNF-a following myocardial injury may activate signaling pathways that promote either cardiac adaptation/protection, or maladaptive responses.
One of the early events in the TNF-a/TNFRl signaling pathways is activation of caspase-8. This is initiated by recruitment of the adaptor protein Fas-associated via a death domain (FADD), which then recruits pro-caspase 8 into the death inducing signaling complex (DISC). Caspase-8 activation leads to the generation of ceramide, mitochondrial reactive oxygen species (ROS) production, Bid cleavage, followed by the Bax-dependent release of cytochrome c from mitochondria, and apoptosome formation, ultimately leading to activation of effectors caspases (i.e. caspase-3) and cell death. In parallel, acute nitric oxide (NO) production through activation of the endothelial (eNOS), or increased expression of inducible nitric oxide synthase (iNOS) inhibits key apoptogenic signals triggered by TNF-a such as ceramide formation and caspase-8. Increased ROS and/or NO-derived reactive species (RNS) change the redox environment of Ca2+ transporters and channels, and thus affect cellular Ca2+ cycling.
[0008] Ryanodine receptors (RyRs) are channels in the sarcoplasmic reticulum (SR) that open and close to regulate the release of Ca2+ from the SR into the intracellular cytoplasm of the cell. The "open probability" (Po) of a RyR refers to the likelihood that the RyR channel is open at any given moment, and therefore capable of releasing Ca2+ into the cytoplasm from the SR. There are three types of ryanodine receptors, RyRl , RyR2, and RyR3, all of which are Ca2+ channels. RyRl is found predominantly in skeletal muscle as well as other tissues, RyR2 is found predominantly in the heart as well as other tissues, and RyR3 is found in the brain as well as other tissues. The RyR channels are formed by four RyR polypeptides in association with four FK506 binding proteins (FKBPs), specifically FKBP12.0 (calstabinl) and FKBP12.6 (calstabin2). Calstabinl binds to RyRl , calstabin2 binds to RyR2, and calstabinl binds to RyR3. The FKBP proteins (calstabinl and calstabin2) bind to the RyR channel (one molecule per RyR subunit), stabilize RyR-channel functioning, and facilitate coupled gating between neighboring RyR channels, thereby preventing abnormal activation of the channel during the channel's closed state.
[0009] The cardiac ryanodine receptor (RyR2), which mediates sarcoplasmic reticulum (SR) Ca2+ release during excitation-contraction coupling, contains about 33 free thiol residues rendering it highly sensitive to the cellular redox state. Cysteine oxidation facilitates RyR opening and SR Ca2+ leak. Moreover, the inventors have recently shown that S-nitrosylation of RyRl (skeletal muscle) and RyR2 (cardiac muscle) and dissociation of the stabilizing subunit calstabinl (FKBP12) or calstabin2 (FKBP12.6) from RyRl and RyR2 complexes respectively induces SR Ca2+ leak, cardiac arrhythmia, skeletal muscle weakness and remodeling in a Duchene muscular dystrophy (mdx) mouse model. However, there is no
report in the literature regarding the role of RyR2 receptor signaling in cardiac ischemia/reperfusion injury.
[0010] As a result of the known roles played by TNF-a signaling in ischemia/reperfusion injury, multicenter trials using TNF-a antagonists in moderate to severe heart failure (HF) demonstrated adverse effects instead of benefits. Thus, a new therapeutic strategy specifically targeting early deleterious effects of TNF-a, without affecting its cytoprotective activity, remains of interest. There is therefore a need in the art for new and improved methods of prevention and treatment of cardiac ischemia/reperfusion injury.
[0011]SUMMARY OF THE INVENTION
[0012] The present invention provides, inter alia, compositions and methods useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury. These compositions and methods involve modulation of the function of cardiac ryanodine receptors.
[0013] The present invention is based, in part, on the discovery that TNF-a-induced caspase-8 activation leads to leaky RyR2 channels that contribute to myocardial remodeling after I/R. More specifically, the applicants have shown that early caspase-8 activation increases mitochondrial ROS and NO production, resulting in S-nitrosylation of RyR2 and depletion of calstabin2 from the channel complex causing a diastolic SR Ca2+ leak that leads to acute pathological left ventricular modeling, and that this can be reversed or prevented by stabilization of the RyR2 macromolecular complex with compounds of formula I as described herein.
[0014] In some preferred embodiments, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, or enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof. The structures of these Formulae are provided in the Detailed Description that follows.
[0015] In a preferred embodiment, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound represented by the structure of Formula I-k as disclosed herein, or enantiomers,
diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof, or any combination thereof.
[0016] In a particularly preferred embodiment, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount a compound represented by the structure of Formula I-o as disclosed hereinor enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof, or any combination thereof.
[0017] In additional preferred embodiments, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of the com ound SI 07 represented by the structure
[0018]
[0019] , or pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof. A preferred salt is the hydrochloride salt (S107-HC1).
[0020] In additional preferred embodiments, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of the compound S36 represented by the structure
[0021]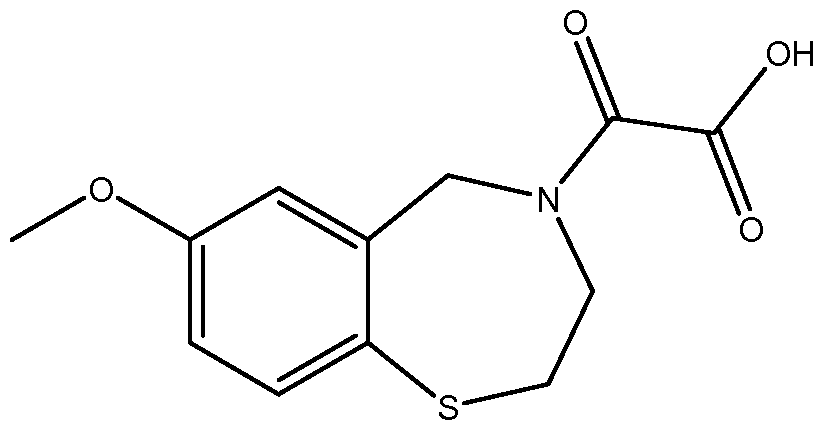 , or pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof. A referred salt is the sodium salt (S36-Na) represented by the structure
, or pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof. A referred salt is the sodium salt (S36-Na) represented by the structure
[0022] In other preferred embodiments, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound represented by the structure of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, 1-h-1, 1-i-1, or Formula II, or enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof.
In other preferred embodiments, the present invention provides a method of treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound represented by the structure of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, 1-h-1, 1-i-1, or Formula II, or enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, or pro-drugs thereof, or any combination thereof.
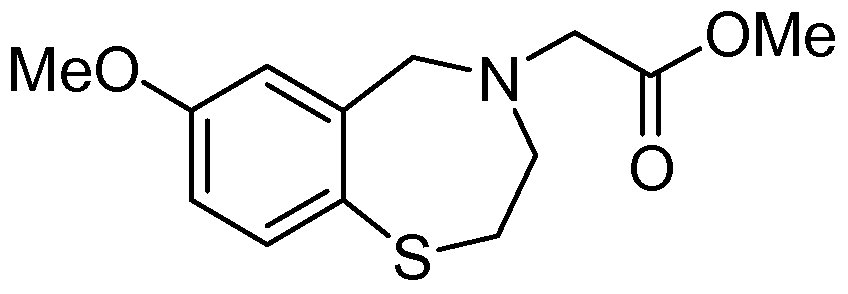
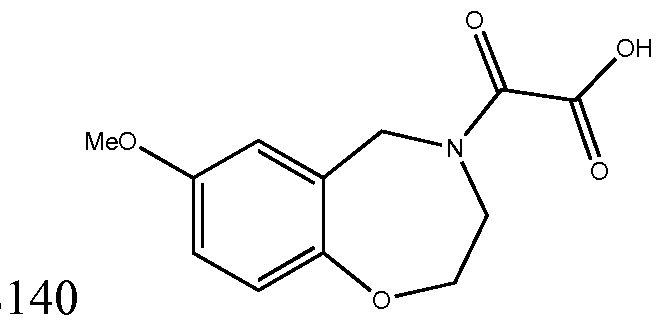
[0023] In certain specific embodiments, the compound administered is selected from the group consisting of SI, S2, S3, S4, S5, S6, S7, S9, SI 1, S12, SI 3, S14, SI 9, S20, S22, S23, S24, S25, S26, S27, S36, S37, S38, S40, S43, S44, S45, S46, S47, S48, S49, S50, S51, S52, S53, S54, S55, S56, S57, S58, S59, S60, S61, S62, S63, S64, S66, S67, S68, S69, S70, S71, S72, S73, S74, S75, S76, S77, S78, S79, S80, S81, S82, S83, S84, S85, S86, S87, S88, S89, S90, S91, S92, S93, S94, S95, S96, S97, S98, S99, S100, S101, S102, S103, S104, S107, S108, S109, S110, Si l l, S112, S113, S114, S115, S116, S117, S118, S119, S120, S121, S122, S123, S136, S137, S138, S139, S140, S146, S147, S148, S149, S150, S151, S152, S153, S156, S157, S159, S160, S161, S166, S167, S182, S186, S189, S203, S217, S251, S252, S258, S277, S279, S282, S291, S293, S296, S301, S302, S306, S311, S312, S313, S318, S322, S324, S326, S331, S335, S337, S351, S352, S353, S354, S397, S398, S399, S423, S454, S463, S466, S470, S473 and S477. The structures of these compounds are provided in the Detailed Description that follows.
[0024] In other embodiments, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound that decreases the open probability of the phosphorylated, and/or nitrosylated, and/or oxidized RyR2 channel under conditions that simulate low activating calcium levels.
[0025] In yet another embodiment, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound that decreases Ca2+ current through the phosphorylated, and/or nitrosylated, and/or oxidized RyR2 channel.
[0026] In a further embodiment, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a
compound that decreases calcium leak through the phosphorylated, and/or nitrosylated, and/or oxidized RyR2 channel under conditions that simulate low activating calcium levels.
[0027] In an additional embodiment, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound that increases the affinity with which calstabin 2 binds to the phosphorylated, and/or nitrosylated, and/or oxidized RyR2.
[0028] In other embodiments, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound that decreases dissociation of calstabin 2 from the phosphorylated, and/or nitrosylated, and/or oxidized RyR2.
[0029] In other embodiments, the present invention provides a method of treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof, comprising administering to the subject a therapeutically or prophylactically effective amount of a compound that increases rebinding of calstabin 2 to the phosphorylated, and/or nitrosylated, and/or oxidized RyR2.
[0030] In these methods, the preferred compounds are those that are specifically described and defined by the formulae disclosed herein.
[0031] In certain embodiments, the subject to whom the compounds of the invention are administered is a mammal selected from the group consisting of primates, rodents, ovine species, bovine species, porcine species, equine species, feline species and canine species. In a preferred embodiment, the subject is a human.
[0032] The compounds of the invention may be administered by any suitable route known in the art, without limitation. For example, compounds of the invention may be administered by a route selected from the group consisting of parenteral, enteral, intravenous, intraarterial, intracardiac, intra intrapericardial, intraosseal, intracutaneous, subcutaneous, intradermal, subdermal, transdermal, intrathecal, intramuscular, intraperitoneal, intrasternal,
[0033]parenchymatous, oral, sublingual, buccal, rectal, vaginal, inhalational, and intranasal.
[0034]Additionally, the compounds of the invention may be administered using a drug-releasing implant.
[0035] In one preferred embodiment, the compounds of the invention are administered to the subject at a dose sufficient to partially or completely restore binding of calstabin 2 to RyR2, or at a dose sufficient to enhance binding of calstabin 2 to RyR2. In certain preferred
embodiments, the compounds of the invention are administered to the subject at a dose of from about 0.01 mg/kg/day to about 20 mg/kg/day, or more preferably still, at a dose of from about 0.05 mg/kg/day to about 1 mg/kg/day. Other suitable dose ranges are provided in the Detailed Description and Examples. In addition, one of skill in the art can select other suitable doses for administration.
[0036] Preferably, the cardiac ischemia/reperfusion injury that is to be trested or prevented in the method of the invention is cardiac ischemia/reperfusion injury following coronary angioplasty or cardiac ischemia/reperfusion injury following thrombolysis during a
[0037]myocardial infarction.
[0038] In other embodiments of the invention, the invention provides use of a compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, I-l, I-m, I-n, I-o, I-p, I-a-1, I-b- 1 , I-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, for preparation of a medicament for treating or preventing treating and/or preventing cardiac ischemia/reperfusion injury in a subject in need thereof. All compounds disclosed herein are expected to be useful for treating or preventing cardiac ischemia/reperfusion injury in a subject in need thereof.
[0039]BRIEF DESCRIPTION OF THE FIGURES
[0040] Figures 1A-G show effect of TNF-a and caspase-8 activation on RyR2 function in vitro.
[0041] Figures 2A-I show roles of caspase-8 and RyR2 leak in myocardial reperfusion injury.
[0042]Figures 3A-C show left ventricular remodeling 15 days after reperfusion.
[0043] Figures 4A-E show the diastolic SR Ca2+ leak via RyR2 channels after
[0044]ischemia/reperfusion contributes to the cardiac remodeling process.
[0045] Figures 5A-C show effect of TNF-a and caspase-8 activation in cardiomyocytes.
[0046]Figures 6A-E show effects of acute TNF-a incubation (10 ng/ml; lh) on Ca2+ transients recorded in fluo-4 AM-loaded intact cardiomyocytes by laser scanning confocal microscopy, in presence of the different caspases inhibitors, the anti-oxidant (NAC) or SI 07.
[0047] Figure 7 shows Bid cleavage assessed by Western blot analysis in sham and I/R hearts after 1 , 6 and 24 hours of reperfusion.
[0048] Figure 8 shows representative sections (Top) of TTC-stained hearts.
[0049] Figures 9A-B show effect of ischemia reperfusion on calstabin2 KO mice.
[0050] Figures lOA-C show effects of TNF-a in WT mice in vivo.
Figures 11A-D show Ca2+ transients recorded in fluo-4 AM-loaded intact
[0051]cardiomyocytes by laser scanning confocal microscopy after 15 days of reperfusion in an in vivo model of ischemia-reperfusion.
[0052] Figure 12 shows representative cardiac RyR2 immunoprecipitation and immunoblots showing the level of PKA phosphorylation level at S2808.
[0053]DETAILED DESCRIPTION OF THE INVENTION
[0054] The following are definitions of terms used in the present specification. The initial definition provided for a chemical group or term herein applies to that group or term throughout the present specification individually or as part of another group, unless otherwise indicated.
[0055] As used herein and in the appended claims, the singular forms "a," "an," and "the" include plural references unless the content clearly dictates otherwise. Thus, for example, reference to "an agent" or "a compound" includes a plurality of such agents or compounds and equivalents thereof known to those skilled in the art.
[0056] As used herein, the term "rycal compounds" refers to compounds of the general Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, 1-b-1, I-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, as provided by the invention, and herein referred to as "compound(s) of the invention".
[0057] The compounds of the invention are referred using a numerical naming system, with compound numbers 1 to 477 provided herein. These numbered compounds are referred to using either the prefix "S." Thus, the first numbered compound is referred to either as "SI", the second numbered compound is referred to as either "S2", the third numbered compound is referred to as either "S3", and so on. The "S" nomenclature systems are used interchangeably throughout the specification, the drawings, and the claims to indicate the specific compounds that are shown by their structures in the Detailed Description.
[0058] The term "alkyl" as used herein refers to a linear or branched, saturated hydrocarbon and preferably one having from 1 to 6 carbon atoms. Representative alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, and neohexyl. The term "C1-C4 alkyl" refers to a straight or branched chain alkane (hydrocarbon) radical containing from 1 to 4 carbon atoms, such as methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, and isobutyl.
[0059] The term "alkenyl" as used herein refers to a linear or branched hydrocarbon and preferably one having from 2 to 6 carbon atoms and having at least one carbon-carbon double
bond. In one embodiment, the alkenyl has one or two double bonds. The double bond may exist as the E or Z isomers and the compounds of the present invention include both isomers.
[0060] The term "alkynyl" as used herein refers to a linear or branched hydrocarbon and preferably one having from 2 to 6 carbon atoms and having at least one carbon-carbon triple bond.
[0061] The term "aryl" as used herein refers to an aromatic group and preferably one containing 1 to 3 aromatic rings, either fused or linked. An example of an aryl group is a phenyl group. The term "cyclic group" as used herein includes a cycloalkyl group and a heterocyclic group.
[0062] The term "cycloalkyl group" as used herein refers to a three- to seven-membered saturated or partially unsaturated carbon ring. Any suitable ring position of the cycloalkyl group may be covalently linked to the defined chemical structure. Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
[0063] The term "halogen" as used herein refers to fluorine, chlorine, bromine, and iodine.
[0064]The term "heterocyclic group" or "heterocyclic" or "heterocyclyl" or "heterocyclo" as used herein refers to fully saturated, or partially or fully unsaturated, including aromatic (i.e., "heteroaryl") cyclic groups (for example, 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 16 membered tricyclic ring systems) which have at least one heteroatom in at least one carbon atom-containing ring. Each ring of the heterocyclic group containing a heteroatom may have 1, 2, 3, or 4 heteroatoms selected from nitrogen atoms, oxygen atoms and/or sulfur atoms, where the nitrogen and sulfur heteroatoms may optionally be oxidized and the nitrogen heteroatoms may optionally be quatemized. The heterocyclic group may be attached to the remainder of the molecule at any heteroatom or carbon atom of the ring or ring system. Examples of heterocyclic groups include, but are not limited to, azepanyl, azetidinyl, aziridinyl, dioxolanyl, furanyl, furazanyl, homo piperazinyl, imidazolidinyl, imidazolinyl, isothiazolyl, isoxazolyl, morpholinyl, oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, piperazinyl, piperidinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazolyl, pyridoimidazolyl, pyridothiazolyl, pyridinyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, quinuclidinyl,
[0065]tetrahydrofuranyl, thiadiazinyl, thiadiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiomorpholinyl, thiophenyl, triazinyl, and triazolyl. Examples of bicyclic heterocyclic groups include indolyl, isoindolyl, benzothiazolyl, benzoxazolyl,
[0066]benzoxadiazolyl, benzothienyl, quinuclidinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuryl, benzofurazanyl,
chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl (such as furo[2,3-c]pyridinyl, furo[3,2-b]pyridinyl] or furo[2,3-b]pyridinyl), dihydroisoindolyl, dihydroquinazolinyl (such as 3,4-dihydro-4-oxo-quinazolinyl),
[0067]triazinylazepinyl, tetrahydroquinolinyl and the like. Examples of tricyclic heterocyclic groups include carbazolyl, benzidolyl, phenanthrolinyl, acridinyl, phenanthridinyl, xanthenyl and the like.
[0068] The term "phenyl" as used herein includes a substituted or unsubstituted phenyl group.
[0069]The aforementioned terms "alkyl," "alkenyl," "alkynyl," "aryl," "acyl," "phenyl," "cyclic group," "cycloalkyl," "heterocyclyl," "heterocyclo," and "heterocycle" may further be optionally substituted with one or more substituents. Examples of substituents include but are not limited to one or more of the following groups: hydrogen, halogen, CF3, OCF3, cyano, nitro, N3, oxo, cycloalkyl, alkenyl, alkynyl, heterocycle, aryl, alkylaryl, heteroaryl, ORa, SRa, S(=0)Re,

[0070]S(=0)2NRbRc,
 C(=0)ORa, C(=0)Ra,
C(=0)ORa, C(=0)Ra,
[0071]NRbC(=0)ORa, NRdC(=0)NRbRe, NRdS(=0)2NRbRc, NRdP(=0)2NRbRc, NRbC(=0)Ra, or NRbP(=0)2Re, wherein Ra is hydrogen, alkyl, cycloalkyl, alkenyl, alkynyl, alkylaryl, heteroaryl, heterocycle, or aryl; Rb, Rc and Rj are independently hydrogen, alkyl, cycloalkyl, alkylaryl, heteroaryl, heterocycle, aryl, or said Rb and Rc together with the N to which they are bonded optionally form a heterocycle; and R» is alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, alkylaryl, heteroaryl, heterocycle, or aryl. In the aforementioned representative substitutents, groups such as alkyl, cycloalkyl, alkenyl, alkynyl, cycloalkenyl, alkylaryl, heteroaryl, heterocycle and aryl can themselves be optionally substituted.
[0072] Representative substituents may further optionally include at least one labeling group, such as a fluorescent, a bioluminescent, a chemiluminescent, a colorimetric or a radioactive labeling group. A fluorescent labeling group can be selected from bodipy, dansyl, fluorescein, rhodamine, Texas red, cyanine dyes, pyrene, coumarins, Cascade Blue™, Pacific Blue, Marina Blue, Oregon Green, 4',6-Diamidino-2-phenylindole (DAPI), indopyra dyes, lucifer yellow, propidium iodide, porphyrins, arginine, and variants and derivatives thereof. For example, SI 18 of the present invention contains a labeling group BODIPY, which is a family of fiuorophores based on the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene moiety. For further information on fluorescent label moieties and fluorescence techniques, see, e.g., Handbook of Fluorescent Probes and Research Chemicals, by Richard P. Haughland, Sixth Edition, Molecular Probes, (1996), which is hereby incorporated by reference in its entirety. One of
skill in the art can readily select a suitable labeling group, and conjugate such a labeling group to any of the compounds of the invention, without undue experimentation.
[0073] The term "quaternary nitrogen" refers to a tetravalent positively charged nitrogen atom including, for example, the positively charged nitrogen in a tetraalkylammonium group (e.g., tetramethylammonium, N-methylpyridinium), the positively charged nitrogen in protonated ammonium species (e.g., trimethyl-hydroammonium, N-hydropyridinium), the positively charged nitrogen in amine N-oxides (e.g., N-methyl-morpholine-N-oxide, pyridine-N-oxide), and the positively charged nitrogen in an N-amino-ammonium group (e.g., N- aminopyridinium) .
[0074] Throughout the specification, unless otherwise noted, the nitrogen in the
[0075]benzothiazepine ring of compounds of the present invention may optionally be a quaternary nitrogen to form, e.g., ammonium derivatives (N(R)4 wherein R is alkyl, aryl, etc.) or
[0076]N-oxides (NO). Non-limiting examples include SI 13 and SI 19.
[0077] The compounds described herein may exist in their tautomeric form (for example, as an amide or imino ether). All such tautomeric forms are contemplated herein as part of the present invention.
[0078] The term "prodrug" as employed herein denotes a compound that, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield compounds of the present invention. For example an ester may be a prodrug of the corresponding carboxylic acid.
[0079] The term "compound(s) of the invention" as used herein means a compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, I-l, I-m, I-n, I-o, I-p, I-a-1, I-b- 1 , I-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, or any of the specific chemical compounds described herein, and salts, hydrates, complexes, metabolites, prodrugs and solvates thereof, or any combination thereof, such as may be used for the treatment or prevention of cardiac ischemia/reperfusion injury.
[0080] A "pharmaceutical composition" refers to a mixture of one or more of the compounds described herein, or pharmaceutically acceptable salts, hydrates or pro-drugs thereof, with other chemical components, such as physiologically acceptable carriers and excipients. The purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism or subject.
[0081] A "pro-drug" refers to an agent which is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they are easier to administer than the parent drug. They are bioavailable, for instance, by oral administration whereas the parent drug is
not. The pro-drug also has improved solubility in pharmaceutical compositions over the parent drug. For example, the compound carries protective groups which are split off by hydrolysis in body fluids, e.g., in the bloodstream, thus releasing active compound or is oxidized or reduced in body fluids to release the compound.
[0082] A compound of the present invention also can be formulated as a pharmaceutically acceptable salt, e.g., acid addition salt, and complexes thereof. The preparation of such salts can facilitate the pharmacological use by altering the physical characteristics of the agent without preventing its physiological effect. Examples of useful alterations in physical properties include, but are not limited to, lowering the melting point to facilitate transmucosal administration and increasing the solubility to facilitate administering higher concentrations of the drug.
[0083] The term "pharmaceutically acceptable salt" means a salt that is suitable for, or compatible with, the treatment of a patient or a subject such as a human patient. The salts can be any non-toxic organic or inorganic salt of any of the compounds represented by Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, 1-b-1, 1-c-1, 1-d-1, I-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II or any of the specific compounds described herein, or any of their intermediates. Illustrative salt-forming ions include, but are not limited to, ammonium (NH4 ), calcium (Ca ), iron (Fe and Fe ), magnesium (Mg ), potassium (K+), pyridinium (C5H5NH+), quaternary ammonium (NR4+), sodium (Na+), acetate, carbonate, chloride, bromide, citrate, cyanide, hydroxide, nitrate, nitrite, oxide, phosphate, sulfate, maleate, fumarate, lactate, tartrate, gluconate, besylate, and valproate. Illustrative inorganic acids that form suitable salts include, but are not limited to, hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as metal salts such as sodium monohydrogen
[0084]orthophosphate and potassium hydrogen sulfate. Illustrative organic acids that form suitable acid addition salts include, but are not limited to, mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene sulfonic and methanesulfonic acids. Either mono or di-acid salts can be formed, and such salts exist in either a hydrated, solvated or substantially anhydrous form. In general, the acid addition salts of compounds of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , I-e-1, 1-f-1, 1-g-1, I-h- 1 , 1-i-1, or Formula II, are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms. The selection of an appropriate salt can be performed by one skilled in the art. For example, one can select salts in reference
to "Handbook of Pharmaceutical Salts : Properties, Selection, and Use" by P. Heinrich Stahl and Camille G. Wermuth, or Berge (1977) "Pharmaceutcial Salts" J. Pharm Sci., Vol 66(1), p 1-19. Other non-pharmaceutically acceptable salts (e.g., oxalates) may be used, for example, in the isolation of compounds of the invention for laboratory use or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
[0085] The compounds of the present invention form hydrates or solvates, which are included in the scope of the claims. When the compounds of the present invention exist as
[0086]regioisomers, configurational isomers, conformers or diasteroisomeric forms all such forms and various mixtures thereof are included in the scope of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, 1-b-1 , 1-c-1, 1-d-1 , 1-e-1, 1-f-1 , 1-g-1 , 1-h-1 , I-i-1 , or Formula II. It is possible to isolate individual isomers using known separation and purification methods, if desired. For example, when a compound of the present invention is a racemate, the racemate can be separated into the (S)-compound and (R)-compound by optical resolution. Individual optical isomers and mixtures thereof are included in the scope of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1 , I-b- 1 , I-c-1 , 1-d-1 , 1-e-1 , I-f- 1 , 1-g-1 , 1-h-1 , I-i-1 , or Formula II.
[0087] The term "solvate" as used herein means a compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1 , 1-b-1 , I-c-1 , 1-d-1 , 1-e-1 , I-f- 1 , I-g-1 , I-h- 1 , I-i-1 , or Formula II, or a pharmaceutically acceptable salt thereof, wherein molecules of a suitable solvent are incorporated in the crystal lattice. A suitable solvent is physiologically tolerable at the dosage administered. Examples of suitable solvents are ethanol, water and the like. When water is the solvent, the molecule is referred to as a "hydrate."
[0088] The terms an "effective amount," "sufficient amount," "therapeutically effective amount," or "prophylactically effective amount" of an agent or compounds, as used herein, refer to amounts sufficient to effect the beneficial or desired results, including clinical results and, as such, the actual "amount" intended will depend upon the context in which it is being applied, such as whether the desired clinical outcome is prevention or treatment. The term "effective amount" also includes that amount of the compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1 , 1-b-1 , I-c-1 , 1-d-1 , 1-e-1 , I-f- 1 , I-g-1 , I-h- 1 , I-i-1 , or Formula II, which is "therapeutically effective" or "prophylactically effective" and which avoids or substantially attenuates undesirable side effects.
[0089] As used herein and as well understood in the art, "treatment" is an approach for obtaining beneficial or desired results, including clinical results. Beneficial or desired clinical
results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state and remission (whether partial or total), whether detectable or undetectable. "Treatment" can also mean prolonging survival as compared to expected survival if not receiving treatment. Unless otherwise stated, the term "treatment" should be construed as encompassing preventive and therapeutic methods.
[0090] The terms "animal," "subject," "organism" and "patient" as used herein include all members of the animal kingdom including, but not limited to, mammals, animals (e.g. , cats, dogs, horses, etc.) and humans.
[0091] All stereoisomers of the compounds of the present invention (for example, those which may exist due to asymmetric carbons on various substituents), including enantiomeric forms and diastereomeric forms, are contemplated within the scope of this invention. Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers (e.g., as a pure or substantially pure optical isomer having a specified activity), or may be admixed, for example, as racemates or with all other, or other selected,
[0092]stereoisomers. The chiral centers of the present invention may have the S or R configuration as defined by the IUPAC 1974 Recommendations. The racemic forms can be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography. The individual optical isomers can be obtained from the racemates by any suitable method, including without limitation, conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization.
[0093] Compounds of the present invention are, subsequent to their preparation, preferably isolated and purified to obtain a composition containing an amount by weight equal to or greater than 99% of the compound ("substantially pure" compound), which is then used or formulated as described herein. Such "substantially pure" compounds of the present invention are also contemplated herein as part of the present invention.
[0094] All configurational isomers of the compounds of the present invention are
[0095]contemplated, either in admixture or in pure or substantially pure form. The definition of compounds of the present invention embraces both cis (Z) and trans (E) alkene isomers, as well as cis and trans isomers of cyclic hydrocarbon or heterocyclic rings.
[0096] Throughout the specification, groups and substituents thereof may be chosen to provide stable moieties and compounds.
The terms "Q-LETD-OPh" and TRP801, referring to a preferential caspase-8 inhibitor, are used interchangeably throughout the specification.
[0097] The term "Q-LETD-OPh" refers to "Na-Quinoline-2-carbonyl-Leu-Glu-Thr- Asp(OMe)-Difluorophenoxymethylketone." The term "Z-IETD-FMK" refers
[0098]"Benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-Fluoromethylketone." The term
[0099]"Z-DEVD-FMK" refers to "Benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)- Fluoromethylketone." The term "Q-VD-OPh" refers to "Quinoline-carbonyl-Val-Asp- Difluorophenoxymethylketone . "
[0100]Prevention and Treatment of Cardiac Ischemia/Reperfusion Injury
[0101] The inventors found that TNF-a induced caspase-8 activation affects RyR2
[0102]S-nitrosylation and leads to diastolic SR Ca2+ leak and left ventricular remodeling in a rat model of ischemia/reperfusion. In particular, the inventors found that early caspase-8 activation increases mitochondrial ROS and NO production, resulting in S-nitrosylation of RyR2 and depletion of calstabin2 (i.e., FKBP12.6) from the channel complex, which causes a diastolic SR Ca2+ leak that leads to acute pathological left ventricular remodeling. This surprising finding identifies RyR2 as a new potential therapeutic target to treat and prevent early myocardial I/R injury.
[0103] Based on these findings, the present invention provides compositions and methods that are useful for treating and/or preventing cardiac ischemia/reperfusion injury. More particularly, the present invention provides compositions comprising the compounds described herein, and methods of treatment and/or prevention comprising administration of these compositions to subjects suffering from, or at risk of developing cardiac
[0104]ischemia/reperfusion injury.
[0105] In certain embodiments, the compositions and methods of the present invention may be used preventively in subjects who are not yet suffering from cardiac ischemia/reperfusion injury, but whom exhibit one or more "risk factors" for cardiac ischemia or are otherwise predisposed to the development of cardiac ischemia, for example, aged individuals, those at risk of ischemic heart disease, and those undergoing clinical procedures involving transient periods of myocardial hypoxia. One example leading to an ischemia/reperfusion event is when blood flow to an ischemic part of the body is restored by surgery, e.g. a bypass of a heart. Another specific example of potential cardiac ischemia/reperfusion injury is heart transplantation. In this procedure, the heart to be transplanted is removed from the donor (ischemic period) and transplanted into the recipient (reperfusion period). The compositon of
the present invention can be given to the recipient to decrease ischemia/reperfusion effects in the body, as well as to be used for fluids to preserve the heart tissue before being transplanted into the recipient. In preferred embodiments of the invention, the cardiac
[0106]ischemia/reperfusion injury that is to be treated or prevented in the method of the invention is cardiac ischemia/reperfusion injury following coronary angioplasty or cardiac
[0107]ischemia/reperfusion injury following thrombolysis during a myocardial infarction.
[0109] In preferred embodiments, the compositions described herein are administered therapeutically or prophylactically to subjects who are suffering from, or at risk of developing cardiac ischemia/reperfusion injury. Such a subject may be any animal that is suffering from, or at risk of developing cardiac ischemia/reperfusion injury. For example, in one
[0110]embodiment, the subject is a mammal. Examples of mammals that may be treated using the methods and compositions of the invention include, but are not limited to, primates, rodents, ovine species, bovine species, porcine species, equine species, feline species and canine species. In preferred embodiments the subjects are human.
[0111] In other embodiments, the "subjects" of the present invention may also be in vitro or in vivo systems, including, without limitation, isolated or cultured cells or tissues, in vitro assay systems.
[0113] The compounds described herein may be formulated into compositions for
[0114]administration to subjects for the treatment and/or prevention of cardiac ischemia/reperfusion injury. The compositions comprise one or more of the benzothiazepine, benzoxazepine, benzodiazepine and benzazepine compounds described herein (such as the compounds of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, 1-b-1, I-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II), in admixture with a
[0115]pharmaceutically acceptable diluent and/or carrier and optionally one or more other pharmarceutically acceptable additives. The pharmaceutically-acceptable diluents and/or carriers and any other additives must be "acceptable" in the sense of being compatible with the other ingredients of the composition and not deleterious to the subject to whom the composition will be administered. One of skill in the art can readily formulate the compounds of the invention into compositions suitable for administration to subjects, such as human subjects, for example using the teaching a standard text such as Remington's Pharmaceutical
Sciences, 18th ed, (Mack Publishing Company: Easton, Pa., 1990), pp. 1635-36), and by taking into account the selected route of delivery.
[0116] Examples of diluents and/or carriers and/or other additives that may be included in the compostions of the invention include, but are not limited to, water, glycols, oils, alcohols, aqueous solvents, organic solvents, DMSO, saline solutions, physiological buffer solutions, peptide carriers, starches, sugars, preservatives, antioxidants, coloring agents, pH buffering agents, granulating agents, lubricants, binders, disintegrating agents, emulsifiers, binders, excipients, extenders, glidants, solubilizers, stabilizers, surface active agents, suspending agents, tonicity agents, viscosity-altering agents, carboxymethyl cellulose, crystalline cellulose, glycerin, gum arabic, lactose, magnesium stearate, methyl cellulose, powders, saline, sodium alginate. The combination of diluents and/or carriers and/or other additives used can be varied taking into account the nature of the active agents used (for example the solubility and stability of the active agents), the route of delivery (e.g. oral, parenteral, etc.), whether the agents are to be delivered over an extended period (such as from a controlled- release capsule), whether the agents are to be co-administered with other agents, and various other factors. One of skill in the art will readily be able to formulate the compounds for the desired use without undue experimentation.
[0117]Dosing & Administration
[0118] In accordance with a method of the present invention, the compounds of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, I-l, I-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , I-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, may be administered to the subject (or contacted with cells of the subject) in an amount effective to treat and/or prevent cardiac
[0119]ischemia/reperfusion injury, and/or in an amount effective to reduce calcium "leak" through the RyR, and/or in an amount effective to reduce the calcium current through the RyR, and/or in an amount effective to stabilize gating of the RyR, and/or in amount effective to increase the binding of calstabin to the RyR complex in the subject, and/or in amount effective to reverse a malfunction of a RyR in the subject, particularly in the cardiac cells of the subject.
[0120] One of skill in the art can readily determine what would be an effective amount of the agents of the invention to be administered to a subject, taking into account whether the agent is being used prophylactically or therapeutically, and taking into account other factors such as the age, weight and sex of the subject, any other drugs that the subject may be taking, any allergies or contraindications that the subject may have, and the like. For example, an effective amount can be determined by the skilled artisan using known procedures, including
analysis of titration curves established in vitro or in vivo. Also, where the desired subject is a human, one of skill in the art can determine the effective dose from performing pilot experiments in suitable animal model species and scaling the doses up or down depending on the subjects weight etc. Effective amounts can also be determined by performing clinical trials in individuals of the same species as the subject, for example starting at a low dose and gradually increasing the dose and monitoring the effects on cardiac ischemia/reperfusion injury. Appropriate dosing regimens can also be determined by one of skill in the art without undue experimentation, in order to determine, for example, whether to administer the agent in one single dose or in multiple doses, and in the case of multiple doses, to determine an effective interval between doses.
[0121] In one embodiment, an effective amount of the compounds of the invention to administer to a subject ranges from about 0.01 mg/kg/day to about 20 mg/kg/day, and/or is an amount sufficient to achieve plasma levels ranging from about 300 ng/ml to about 1000 ng/ml. In one embodiment, the amount of compounds from the invention ranges from about 5 mg/kg/day to about 20 mg/kg/day. In another embodiment, from about 10 mg/kg/day to about 20 mg/kg/day is administered. In another embodiment, from about 0.01 mg/kg/day to about 10 mg/kg/day is administered. In another embodiment, from about 0.01 mg/kg/day to about 5 mg/kg/day is administered. In another embodiment, from about 0.05 mg/kg/day to about 5 mg/kg/day is administered. In another, preferred embodiment, from about 0.05 mg/kg/day to about 1 mg/kg/day is administered.
[0122] The compositions described herein may be administered to a subject by any suitable method that allows the agent to exert its effect on the subject in vivo. For example, the compositions may be administered to the subject by known procedures including, but not limitated to, by oral administration, sublingual or buccal administration, parenteral
[0123]administration, transdermal administration, via inhalation, via nasal delivery, vaginally, rectally, and intramuscularly. The compounds of the invention may be administered parenterally, or by epifascial, intracapsular, intracutaneous, subcutaneous, intradermal, intrathecal, intramuscular, intraperitoneal, intrasternal, intravascular, intravenous,
[0124]parenchymatous, or sublingual delivery. Delivery may be by injection, infusion, catheter delivery, or some other means, such as by tablet or spray. In one embodiment, the agent is adiminstered to the subject by way of delivery directly to the heart tissue, such as by way of a catheter inserted into, or in the proximity of the subject's heart, or by using delivery vehicles capable of targeting the drug to the heart. For example, the compounds of the invention may
be conjugated to or administered in conjunction with an agent that is targeted to the heart, such as an antibody or antibody fragment.
[0125] For oral administration, a formulation of the compounds of the invention may be presented as capsules, tablets, powders, granules, or as a suspension or solution. The formulation may contain conventional additives, such as lactose, mannitol, cornstarch or potato starch, binders, crystalline cellulose, cellulose derivatives, acacia, cornstarch, gelatins, disintegrators, potato starch, sodium carboxymethylcellulose, dibasic calcium phosphate, anhydrous or sodium starch glycolate, lubricants, and/or or magnesium stearate.
[0126] For parenteral administration (i.e., administration by through a route other than the alimentary canal), the compounds of the invention may be combined with a sterile aqueous solution that is isotonic with the blood of the subject. Such a formulation may be prepared by dissolving the active ingredient in water containing physiologically-compatible substances, such as sodium chloride, glycine and the like, and having a buffered pH compatible with physiological conditions, so as to produce an aqueous solution, then rendering the solution sterile. The formulation may be presented in unit or multi-dose containers, such as sealed ampoules or vials. The formulation may be delivered by injection, infusion, or other means known in the art.
[0127] For transdermal administration, the compounds of the invention may be combined with skin penetration enhancers, such as propylene glycol, polyethylene glycol, isopropanol, ethanol, oleic acid, N-methylpyrrolidone and the like, which increase the permeability of the skin to the compounds of the invention and permit the compounds to penetrate through the skin and into the bloodstream. The compositions also may be further combined with a polymeric substance, such as ethylcellulose, hydroxypropyl cellulose, ethylene/vinylacetate, polyvinyl pyrrolidone, and the like, to provide the composition in gel form, which are dissolved in a solvent, such as methylene chloride, evaporated to the desired viscosity and then applied to backing material to provide a patch.
[0128] In some embodiments, the composition is in unit dose form such as a tablet, capsule or single-dose injection or infusion vial.
[0129] In certain embodiments, the agents described herein may be used in combination with other agents useful for the treatment of cardiac ischemia/reperfusion injury or with other agents that ameliorate the effect of certain risk factors for cardiac ischemia/reperfusion injury. For example, in one embodiment, the agents of the invention may be delivered to a subject as part of a composition containing one or more additional active agents. In another
[0130]embdodiment, the agents of the invention may be delivered to a subject in a composition or
formulation containing only that active agent, while one or more other agents useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury may also be administered to the subject in one or more separate compositions or formulations.
[0131] The agents of the invention and the other agents useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury may be administered to the subject at the same time, or at different times. For example, the agents of the invention and the other agents may be administered within minutes, hours, days, weeks, or months of each other, for example as part of the overall treatment regimen of a subject.
[0132] Agents of the invention useful for treating and/or preventing cardiac
[0133]ischemia/reperfusion injury may be used in combination with the other agents that include, but are not limited to, β-adrenergic blockers, calcium channel blockers and anti-arrhythmic drugs.
[0134] When the treatment follows a myocardiac infarction, the compounds of the invention should be administered within 2-4 hours of the onset of symptoms, prior to or simultaneously with the reperfusion therapy. In one embodiment, a compound of the invention is
[0135]administered together with the drugs for thromblytic therapy. In another embodiment, a compound of the invention is administered during percutaneous coronary intervention (PCI). The use of percutaneous coronary intervention as a therapy to abort a myocardial infarction is known as primary PCI. The goal of primary PCI is to open the artery as soon as possible, and preferably within 90 minutes of the patient presenting to the emergency room. Primary PCI involves performing a coronary angiogram to determine the anatomical location of the infarcting vessel, followed by balloon angioplasty (and frequently deployment of an intracoronary stent) of the thrombosed arterial segment. In a preferred embodiment, when an intracoronary stent is used for PCI, the compound of the invention may be provided on the stent in a conventional manner, such as by coating.
[0136] The present compounds are expected to prevent the ischemia/reperfusion injury, have a high sustained patency rate, are easily and rapidly administered, and have no antigenicity or adverse hemodynamic effects, or no known clinically significant drug interactions.
[0137] In yet another embodiment, a compound of the invention is administered during bypass surgery. Emergency bypass surgery for the treatment of an acute myocardial infarction (MI) is less common than PCI or medical management. Emergency coronary artery bypass graft surgery (CABG) is usually undertaken to simultaneously treat a mechanical complication, such as a ruptured papillary muscle, or a ventricular septal defect, with ensueing cardiogenic shock.
The administration of a compounds of the invention in connection with this treatment is expected to lead to optimal results. Typically the heart is stopped during surgery and the blood is circulated mechanically. The compound can be administered by adding an appropriate dosage into the recirculated blood or by directly administering the compound in a suitable solution into the heart or open blood vessel.
[0138]Screening for New Compounds Useful for Treating Cardiac Ischemia/reperfusion Injury
[0139] In another embodiment, the present invention is directed to methods for identifying additional compounds that may be useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury. Such methods may be based on, inter alia, identifying compounds that increase binding of calstabins to RyRs, and/or decrease the calcium current through RyR channels, and the like. Examples of suitable assays and screening methods that may be used to identify new compounds that may be useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury are described in U.S. patent applications 09/568,474, 10/288,606, 10/680,988, 10/608,723, 10/809,089, 10/763,498, 10/794,218, 11/088,058, 11/088,123, 11/212,309, 11/506,285, and 11/212,413, the entire contents of each of which are hereby incorporated by reference.
[0141] The present invention encompasses compounds useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury, and methods of treatment and/or prevention comprising administration of such compounds, or compositions containing such compounds, to subjects who are suffering from, or who are at risk of developing, cardiac
[0142]ischemia/reperfusion injury. The compounds of the invention indirectly decrease the open probability of RyR when examined under conditions that simulate diastole, by inhibiting the depletion of the stabilizing subunit calstabin2 from the RyR2 complex and thereby stabilizing the closed state of the channel, particularly protein kinase A (PKA) phosphorylated, and/or nityrosylated, and/or oxidized RyR, and thereby decrease the Ca2+ current through such channels under resting conditions when muscles are relaxed. The compounds of the invention exert this effect, at least in part, by increasing the affinity with which calstabin proteins bind to RyRs, and/or by inhibiting a decrease in binding of calstabins to RyRs, and/or by inhibiting dissociation of calstabins from RyRs, particularly PKA phosphorylated RyRs. The compounds of the invention decrease the open probability of RyR and decrease the "leak" of
Ca2+ through such channels by stabilizing the closed state of the channel without blocking the channel pore.
[0143] The present invention relates to use of benzothiazepine, benzoxazepine,
[0144]benzodiazepine and benzazepine compounds in the treatment and/or prevention of cardiac ischemia/reperfusion injury. In preferred embodiments, the present invention provides benzothiazepine, benzoxazepine, benzodiazepine and benzazepine compounds as described by the chemical Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1, I-b- 1 , 1-c-1, I-d- 1 , 1-e-1, I-f- 1 , 1-g-1, I-h- 1 , I-i- 1 , or Formula II, as described below.
[0145] In one aspect, the present invention provides methods for the treatment and/or prevention of cardiac ischemia/reperfusion injury that comprise administering compounds of Formula I to subjects in need thereof. In another aspect, the present invention provides compositions useful for the treatment and/or prevention of cardiac ischemia/reperfusion injury that comprise compounds of Formula I. The structure of Formula I is as follows:
[0146] ormula I) wherein,
ormula I) wherein,
[0147] T is O, CH2, NH, or S=(02)n;
[0149]q is 0, 1, 2, 3, or 4;
[0150]each R is independently selected from the group consisting of H, halogen, -OH, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, -O-acyl, alkyl, alkoxyl, alkylamino, alkylarylamino, alkylthio, cycloalkyl, alkylaryl, aryl, heteroaryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and
[0151](hetero-)arylamino; wherein each acyl, -O-acyl, alkyl, alkoxyl, alkylamino, alkylarylamino, alkylthio, cycloalkyl, alkylaryl, aryl, heteroaryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino may be optionally substituted;
Ri is selected from the group consisting of H, oxo, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein each alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl may be optionally substituted;
[0152] R2 is selected from the group consisting of H, -C(=0)R5, -C(=S)R6, -S02R7, -P(=0)R8R9, -(CH2)m-Rio, alkyl, aryl, alkylaryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl; wherein each alkyl, aryl, alkylaryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl may be optionally substituted and wherein m is 0, 1, 2, 3, or 4;
[0153] R3 is selected from the group consisting of H, -C02Y, -C(=0)NHY, acyl, -O-acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl may be optionally substituted; and wherein Y is selected from the group consisting of H, alkyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl, and wherein each alkyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl may be optionally substituted;
[0154] R4 is selected from the group consisting of H, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein each alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, heteroaryl, and heterocyclyl may be optionally substituted;
[0155]R5 is selected from the group consisting of -NR15R16, -(CH2)zNRi5Ri6, -NHNR15R16, -NHOH, -ORis, -C(=0)NHNRi5Ri6, -C02Ri5,
 -CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6;
-CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6;
[0156]Re is selected from the group consisting of -OR15, -NHNR15R16, -NHOH, -NR15R16, -CH2X, acyl, alkenyl, alkyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkyl, aryl, alkylaryl, cycloalkyl,
[0157]cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted;
[0158]R7 is selected from the group consisting of -OR15, -NR15R16, -NHNR15R16, -NHOH, -CH2X, alkyl, alkenyl, alkynyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each alkyl, alkenyl, alkynyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted;
[0159]Rg and R9 independently are selected from the group consisting of OH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, alkylaryl,
cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted;
[0160] Rio is selected from the group consisting of -NR15R16, OH, -S02Rn, -NHS02Rn, C(=0)(Ri2), NHC=0(Ri2), -OC=0(Ri2), and -P(=0)Ri3Ri4;
[0161] R11, Ri2, Ri3, and R14 independently are selected from the group consisting of H, OH, NH2, -NHNH2, -NHOH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl,
[0162]heterocyclyl, and heterocyclylalkyl may be optionally substituted;
[0163] X is selected from the group consisting of halogen, -CN, -C02Ri5, -C(=0)NR15R16, -NR15R16,

[0164] Ri5 and Ri6 independently are selected from the group consisting of H, acyl, alkenyl, alkoxyl, OH, NH2, alkyl, alkylamino, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted; and optionally Ri5 and Ri6 together with the N to which they are bonded may form a heterocycle which may be substituted;
[0165]the nitrogen in the benzothiazepine ring may optionally be a quaternary nitrogen; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, and prodrugs thereof.
[0166] Examples of compounds that may be used in conjunction with the invention include, without limitation, SI, S2, S3, S4, S5, S6, S7, S9, SI 1, S12, SI 3, S14, SI 9, S20, S22, S23, S24, S25, S26, S27, S36, S37, S38, S40, S43, S44, S45, S46, S47, S48, S49, S50, S51, S52, S53, S54, S55, S56, S57, S58, S59, S60, S61, S62, S63, S64, S66, S67, S68, S69, S70, S71, S72, S73, S74, S75, S76, S77, S78, S79, S80, S81, S82, S83, S84, S85, S86, S87, S88, S89, S90, S91, S92, S93, S94, S95, S96, S97, S98, S99, S100, S101, S102, S103, S104, S107, S108, S109, S110, Si l l, S112, S113, S114, S115, S116, S117, S118, S119, S120, S121, S122, S123, S136, S137, S138, S139, S140, S146, S147, S148, S149, S150, S151, S152, S153, S156, S157, S159, S160, S161, S166, S167, S182, S186, S189, S203, S217, S251, S252, S258, S277, S279, S282, S291, S293, S296, S301, S302, S306, S311, S312, S313, S318, S322, S324, S326, S331, S335, S337, S351, S352, S353, S354, S397, S398, S399,
S423, S454, S463, S466, S470, S473 and S477, as herein defined. In certain embodiments, the compounds are isolated and substantially pure.
[0167] In one embodiment, the present invention provides methods and uses which comprise administering compounds of Formula I-a':
[0168]
[0170] T is O, CH2, NH, or S=(02)n;
[0172]q is 0, 1 , 2, 3, or 4; each R is independently selected from the group consisting of H, halogen, -OH, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; wherein each acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino may be substituted or unsubstituted;
[0173]R2 is selected from the group consisting of H, -C=0(R5), -C=S(Re), -S02R7, -P(=0)R8R9, -(CH2)m-Rio, alkyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl; wherein each alkyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl may be substituted or unsubstituted, wherein m is 0, 1 , 2, 3, or 4;
[0174]R5 is selected from the group consisting of -NR15R16, -(CH2)zNRi5Ri6, -NHNR15R16, -NHOH, -ORis, -C(=0)NHNRi5Ri6, -C02Ri5,
 -CH2X, acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted, and wherein z is 1 , 2, 3, 4, 5, or 6;
-CH2X, acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted, and wherein z is 1 , 2, 3, 4, 5, or 6;
[0175]Re is selected from the group consisting of -OR15, -NHNR15R16, -NHOH, -NR15R16, -CH2X, acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
R7 is selected from the group consisting of H, -OR15, -NR15R16, -NHNR15R16, -NHOH, -CH2X, alkyl, akenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and
[0176]heterocyclylalkyl; wherein each alkyl, akenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0177] Rg and R9 independently are selected from the group consisting of -OH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl;
[0178]wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0179] Rio is selected from the group consisting of -NR15R16, OH, -S02Rn, -NHS02Rn, -C(=0)Ri2, -NH(C=0)Ri2, -0(C=0)Ri2, and -P(=0)Ri3Ri4;
[0180] R11, R12, Ri3, and Ri4 independently are selected from the group consisting of H, OH, NH2, -NHNH2, -NHOH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0181] X is selected from the group consisting of halogen, -CN, -C02Ris,
 -NRi5Ri6,
-NRi5Ri6,

[0182] Ri5 and Ri6 independently are selected from the group consisting of H, acyl, alkenyl, alkoxyl, OH, NH2, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and
[0183]heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted; and optionally R15 and R½ together with the N to which they are bonded may form a heterocycle which may be substituted or unsubstituted;
[0184]the nitrogen in the benzothiazepine ring may be optionally a quaternary nitrogen; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, and prodrugs thereof.
[0185] In one embodiment, the present invention provides methods and uses which comprise administering compounds of Formula I-a:

[0188]q is 0, 1, 2, 3, or 4;
[0189]each R is independently selected from the group consisting of H, halogen, -OH, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; wherein each acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino may be substituted or unsubstituted;
[0190]R2 is selected from the group consisting of H, -C=0(R5), -C=S(R6), -S02R7, -P(=0)R8R9, -(CH2)m-Rio, alkyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl; wherein each alkyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl, and heterocyclyl may be substituted or unsubstituted, wherein m is 0, 1, 2, 3, or 4;
[0191] R5 is selected from the group consisting of -NRi5Ri6, -(CH2)zNRi5Ri6, -NHNRi5Ri6, -NHOH, -ORis, -C(=0)NHNRi5Ri6, -C02Ri5,
 -CH2X, acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted, and wherein z is 1, 2, 3, 4, 5, or 6;
-CH2X, acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted, and wherein z is 1, 2, 3, 4, 5, or 6;
[0192]Re is selected from the group consisting of -ORi5, -NHNR15R16, -NHOH, -NR15R16, -CH2X, acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0193]R7 is selected from the group consisting of H, -OR15, -NR15R16, -NHNR15R16, -NHOH, -CH2X, alkyl, akenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and
heterocyclylalkyl; wherein each alkyl, akenyl, alkynyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0194] Rg and R9 independently are selected from the group consisting of -OH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl;
[0195]wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0196] Rio is selected from the group consisting of -NR15R16, OH, -S02Rn, -NHS02Rn, -C(=0)Ri2, -NH(C=0)Ri2, -0(C=0)Ri2, and -P(=0)Ri3Ri4;
[0197] R11, R12, Ri3, and R14 independently are selected from the group consisting of H, OH, NH2, -NHNH2, -NHOH, acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0198] X is selected from the group consisting of halogen, -CN, -CO2R15, -C(=0)NR15R16, -NR15R16,

[0199] Ri5 and Ri6 independently are selected from the group consisting of H, acyl, alkenyl, alkoxyl, OH, NH2, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and
[0200]heterocyclylalkyl; wherein each acyl, alkenyl, alkoxyl, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted; and optionally R15 and R½ together with the N to which they are bonded may form a heterocycle which may be substituted or unsubstituted;
[0201]the nitrogen in the benzothiazepine ring may be optionally a quaternary nitrogen; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, and prodrugs thereof.
[0202] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-a, wherein each R is independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph,
[0203]-C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1, or 2.
[0204] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-a, wherein R2 is selected from the group
consisting of-C=0(R5), -C=S(R6), -S02R7, -P(=0)R8R9, and -(CH2)m-Rio, wherein m is 0, 1, 2, 3, or 4.
[0205] In yet another embodiment, the present invention provides methods and uses which comprise administering com ounds of formula I-b:
[0206]
[0207]wherein R and R" are independently selected from the group consisting of H, halogen, -OH, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0208] R2 and n are as defined in compounds of formula I-a above;
[0209]and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0210] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-b, wherein R and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R is H or OMe, and R" is H.
[0211] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-b, wherein R2 is selected from the group consisting of-C=0(R5), -C=S(R6), -S02R7, -P(=0)R8R9, and -(CH2)m-Ri0.
[0212] In yet another embodiment, the present invention provides methods and uses which comprise administering compounds formula of I-c:

[0213]wherein each R, R7, q, and n is as defined in compounds of formula I-a above; and
[0214]enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, metabolites, and pro-drugs thereof.
[0215] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-c, wherein each R is independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)C C4alkyl, -S-C C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph,
[0216]-C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1, or 2.
[0217] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-c, wherein R7 is selected from the group consisting of -OH, -NRi5Ri6, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each alkyl, akenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted.
[0218] In a further embodiment, the present invention provides methods and uses which comprise administering com ounds of formula of I-d:
[0219]
[0220]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0221]-NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl,
alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0222] R7 and n are as defined in compounds of formula I-a above; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0223] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-d, wherein R and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R is H or OMe, and R" is H.
[0224] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-d, wherein Ry is selected from the group consisting of -OH, -NR15R16, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each alkyl, akenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted.
[0225] In one embodiment, the present invention provides methods and uses which comprise administering compounds of formula of I-e:
[0226]
[0227]wherein each R, R5, q and n is as defined compounds of formula I-a above; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0228] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-e, wherein each R is independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3,
-S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph,
[0229]-C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1, or 2.
[0230] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-e, wherein R5 is selected from the group consisting of-NRi5Ri6, -(CH2)zNRi5Ri6, -NHOH, -ORi5, -CH2X, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted.
[0231] In some embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-e, wherein R5 is an alkyl substituted by at least one labeling group, such as a fluorescent, a bioluminescent, a chemiluminescent, a colorimetric and a radioactive labeling group. A fluorescent labeling group can be selected from bodipy, dansyl, fluorescein, rhodamine, Texas red, cyanine dyes, pyrene, coumarins, Cascade Blue™, Pacific Blue, Marina Blue, Oregon Green, 4',6-Diamidino-2-phenylindole (DAPI), indopyra dyes, lucifer yellow, propidium iodide, porphyrins, arginine, and variants and derivatives thereof.
[0232] In another embodiment, the present invention provides methods and uses which comprise administering compounds of formula of I-f:
[0233] (i-f)
(i-f)
[0234]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0235]-NH2, -NO2, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0236] R5 and n are as defined in compounds of formula I-a above;
and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0237] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-f, wherein R' and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R is H or OMe, and R" is H.
[0238] A preferred compound of formula I-f is S36, in particular in the form of a sodium salt.
[0239]In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-f, wherein -(CH2)zNRi5Ri6, selected from the group consisting of-NRi5Ri6, -NHOH, -ORi5, -CH2X, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted.
[0240] In yet another embodiment, the present invention provides methods and uses which comprise administerin mpounds of formula of I-g:
[0241]
[0242]wherein W is S or O; each R, Ri5, Ri6, q, and n is as defined in compounds of formula I-a above; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0243] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-g, wherein each R is independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1, or 2.
In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-g, wherein Ri5 and Ri6 independently are selected from the group consisting of H, OH, NH2, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted; and optionally R15 and R½ together with the N to which they are bonded may form a heterocycle which may be substituted.
[0244] In some embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-g, wherein W is O or S.
[0245] In yet another embodiment, the present invention provides methods and uses which comprise administerin compounds of formula of I-h:
[0246]
[0247]wherein W is S or O;
[0248]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0249]-NH2, -NO2, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0250] Ri5, Ri6 and n are as defined in compounds of formula I-a above;
[0251]and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0252] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-h, wherein R and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3,
-N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R' is H or OMe, and R" is H.
[0253] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-h, wherein R15 and Ri6 independently are selected from the group consisting of H, OH, NH2, alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each alkyl, alkylamino, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted; and optionally R15 and R½ together with the N to which they are bonded may form a heterocycle which may be substituted.
[0254] In some embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-h, wherein W is O or S.
[0255] In a further embodiment, the present invention provides methods and uses which comprise administerin compounds of formula of I-i:
[0256]
[0257]wherein Rn is selected from the group consisting of -NRi5Ri6, -NHNRi5Ri6, -NHOH, -OR15, -CH2X, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl;
[0258]wherein each alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0259]each R, q, and n is as defined in compounds of formula I-a above; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0260] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-i, wherein each R is independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3,
-S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph,
[0261]-C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1, or 2.
[0262] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-i, wherein Rn is -NRi5Ri6, and -ORi5. In certain other embodiments, Ri7 is -OH, -OMe, -NEt, -NHEt, -NHPh, -NH2, or
[0264] In one embodiment, the present invention provides methods and uses which comprise administering compounds of formula of I- :
[0265]
[0266]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0267]-NH2, -N02, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0268] Ri7 is selected from the group consisting of -NRi5Ri6, -NHNRi5Ri6, -NHOH, -ORi5, -CH2X, alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each alkenyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted;
[0269]n is as defined in compounds of formula I-a; and
[0270]enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0271] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-j, wherein R and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3,
-N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R' is H or OMe, and R" is H.
[0272] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-j, wherein R17 is -NR15R16 or -OR15. In certain other embodiments, Rn is -OH, -OMe, -NEt, -NHEt, -NHPh, -NH2, or
[0274] In another embodiment, the present invention provides methods and uses which comprise administering compounds of formula I-k:
[0275] (I-k)
(I-k)
[0276]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0277]-NH2, -NO2, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0278] Ri5 and Ri6 are as defined in Formula (I),
[0279]Ris is selected from the group consisting of -NRi5Ri6,
 -(C=0)OR15, -OR15, alkyl, aryl, cycloalkyl, heterocyclyl, and at one labeling group; wherein each alkyl, aryl, cycloalkyl, and heterocyclyl may be substituted or unsubstituted;
-(C=0)OR15, -OR15, alkyl, aryl, cycloalkyl, heterocyclyl, and at one labeling group; wherein each alkyl, aryl, cycloalkyl, and heterocyclyl may be substituted or unsubstituted;
[0280]wherein p is any one of 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10, with each value of p representing a different embodiment;
[0281]and n is 0, 1, or 2;
[0282]and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
In certain preferred embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-k, wherein R' and R" are
[0283]independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)C C4alkyl, -S-C C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, C2-C4alkoxyl, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R' is H or OMe, and R" is H.
[0284] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-k, wherein Ri8 is selected from the group consisting of-NRi5Ri6, -(C=0)OR15, -OR15, alkyl, aryl, and at one labeling group; and wherein each alkyl and aryl may be substituted or unsubstituted. In some cases, n is 1, and Ri8 is Ph, C(=0)OMe, C(=0)OH, aminoalkyl, NH2, NHOH, or NHCbz. In other cases, n is 0, and Rig is Ci-C4 alkyl, such as Me, Et, propyl, and butyl. In yet other cases, n is 2, and Ri8 is pyrrolidine, piperidine, piperazine, or morpholine. In some embodiments, m is 3, 4, 5, 5, 7, or 8, and Rig is a fluorescent labeling group selected from bodipy, dansyl, fluorescein, rhodamine, Texas red, cyanine dyes, pyrene, coumarins, Cascade Blue™, Pacific Blue, Marina Blue, Oregon Green, 4',6-Diamidino-2-phenylindole (DAPI), indopyra dyes, lucifer yellow, propidium iodide, porphyrins, arginine, and variants and derivatives thereof.
[0285] In some embodiments, Rig in formula I-k is selected from the group consisting of -NR15R16, -(C=0)ORi5, -OR15, alkyl, and aryl, wherein each alkyl and aryl may be substituted or unsubstituted. Most preferably, in Formula I-k, R is H, OMe, or C2-C4alkoxyl; R" is H; n is 0; and Rig is Ci-C4alkyl.
[0286] In yet another embodiment, the present invention provides methods and uses which comprise administering com ounds of formula of I-l:
[0287]
[0288]wherein R and R" are independently selected from the group consisting of H, halogen, -OH,
[0289]-NH2, -NO2, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl,
alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0290]Re and n are as defined in compounds of formula I-a; and enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0291] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula 1-1, wherein R' and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R' is H or OMe, and R" is H.
[0292] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula 1-1, wherein R^ is selected from the group consisting of -NRi5Ri6, -NHNRi5Ri6, -ORi5, -NHOH, -CH2X, acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkenyl, alkyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted or unsubstituted. In some cases, Re is -NR15R16 such as -NHPh, pyrrolidine, piperidine, piperazine, morpholine, and the like. In some other cases, R6 is alkoxyl, such as -O-tBu.
[0293] In a further embodiment, the present invention provides methods and uses which comprise administering com ounds of formula I-m:
[0294]
[0295]wherein R' and R" are independently selected from the group consisting of H, halogen, -OH,
[0296]-NH2, -NO2, -CN, -CF3, -OCF3, -N3, -S03H, -S(=0)2alkyl, -S(=0)alkyl, -OS(=0)2CF3, acyl, alkyl, alkoxyl, alkylamino, alkylthio, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl,
alkynyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; and wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, aryl, heterocyclyl, heterocyclylalkyl, alkenyl, alkynyl, (hetero-)aryl, (hetero-)arylthio may be substituted or unsubstituted;
[0297] Rg, R9 and n are as defined in compounds of formula I-a above; and enantiomers,
[0298]diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof.
[0299] In certain embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-m, wherein R and R" are independently selected from the group consisting of H, halogen, -OH, OMe, -NH2, -N02, -CN, -CF3, -OCF3, -N3, -S(=0)2Ci-C4alkyl, -S(=0)Ci-C4alkyl, -S-Ci-C4alkyl, -OS(=0)2CF3, Ph, -NHCH2Ph, -C(=0)Me, -OC(=0)Me, morpholinyl and propenyl; and n is 0, 1 or 3. In some cases, R is H or OMe, and R" is H.
[0300] In other embodiments, the present invention provides methods and uses which comprise administering compounds of formula I-m, wherein Rg and R9 are independently alkyl, aryl, -OH, alkoxyl, or alkylamino. In some cases, Rg is Ci-C4 alkyl such as Me, Et, propyl and butyl; and R9 is aryl such as phenyl.
[0301] In other embodiments, the present invention provides methods and uses which comprise administering com ounds of formula I-n,
[0302]
[0305]Rd is CH2, or NRa; and
[0306] Ra is H, -(Ci-C6 alkyl)-aryl, wherein the aryl is a disubstituted phenyl or a
[0307]benzo[l,3]dioxo-5-yl group, or an amine protecting group (e.g., a Boc group); and
[0308] Rb is hydrogen of alkoxy (e.g., methoxy).
[0309] Representative compounds of Formula I-n include without limitation S101, SI 02,
[0310]S103, S114.
In certain other embodiments, the invention provides compounds of Formula I-o:
[0311]
[0314]Re is substituted or unsubstituted -Ci-C6 alkyl, -(Ci-C6 alkyl)-phenyl, or -(Ci-C6 alkyl)-C(0)Rb; and
[0315] Rb is -OH or -0-(Ci-C6 alkyl), and
[0316] wherein the phenyl or substituted alkyl is substituted with one or more of halogen, hydroxyl, -Ci-C6 alkyl, -0-(Ci-C6 alkyl), -NH2, -NH(Ci-C6 alkyl), -N(Ci-C6 alkyl)2, cyano, or dioxolane.
[0317] Representative compounds of Formula I-o include without limitation S 107, SI 10, Si l l, S120, and S121.
[0318] In certain other embodiments, the invention provides compounds of Formula I-p:
[0319]
[0322] Rc is -(Ci-C6 alkyl)-NH2, -(C C6 alkyl)-ORf, wherein Rf is H or -C(0)-(Ci-C6)alkyl, or -(Ci-C6 alkyl)-NHRg wherein Rg is carboxybenzyl. Representative compound of Formula I-p include without limitation SI 09, S122, and SI 23.
[0323] The compounds of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, and Formula II can be used in methods that treat and/or prevent cardiac ischemia/reperfusion injury, and may also be used in compositions suitable for the treatment and/or prevention of cardiac ischemia/reperfusion injury. In one preferred embodiment, the
compounds used have structures as described by Formula I-a, I-b, I-e, I-f, I-g, I-h, I-i, I-j, I-k, I-n, I-o, or I-p.
[0324] Another preferred embodiment relates to compounds of Formula I-a-1 :
[0325] wherein
wherein
[0326] n is 0, 1, 2, 3, or 4;
[0327] each R is independently selected from the group consisting of Z, R5, -OR5, -SR5, -N(R5)2, -NR5C(=0)OR5, -C(=0)N(R5)2, -C(=0)OR5, -C(=0)R5, -OC(=0)R5, N02, CN, -CZ3, OCZ3, -N3, and -P(=0)R8R9;
[0328] Ri and R3 are each independently selected from the group consisting of oxo, R5, -CH2OR5, -CH2OC(=0)R6, -C(=0)OR5, -C(=0)NHR5, -C(=0)R5, and -OC(=0)R5;
[0329]R2 is selected from the group consisting of R5, -C(=0)R6, -C(=S)R6, and -(CH2)mRi0, wherein m is 1, 2, 3, 4, 5, or 6; or
[0330] Ri and R2 together with the carbon and nitrogen to which they are respectively attached, form an unsubstituted or substituted heterocycle; or
[0331] R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsubstituted or substituted heterocycle other than a piperazine; or
[0332]R3 and R4 together with the carbon atoms to which they are respectively attached, form an unsubstituted or substituted cycloalkyl or heterocyclic ring; or
[0333] R4 is selected from the group consisting of R5 and oxo;
[0334] each R5 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, alkylaryl, and alkylheteroaryl;
Re is selected from the group consisting of R5, -(CH2)bNRi3Ri4, -NR5OR5, -OR5, -C(=0)OR5, -C(=0)NRi3Ri4, -(CH2 )cY, and -C(=0)R5, wherein b is 0, 1 , 2, 3, 4, 5, or 6 and c is 1 , 2, 3, 4 or 5;
[0335]Rio is selected from the group consisting of R5, -OR5, -S02Rn, -C(=0)Ri2,
[0336] -NH(C=0)Ri2, -0(C=0)Ri2, and -P(=0)R8R9;
[0337] Rg, R9, R11 and Ri2 are independently selected from the group consisting of R5, OR5, and -N(R5)2;
[0338]Y is selected from the group consisting of Z, -C02R5, -C(=0)NRi3Ri4, and -OR5; Z is a halogen selected from F, CI, Br and I;
[0339]Ri3 and Ri4 are independently selected from the group consisting of R5, or Ri3 and Ri4 together with the N to which they are bonded may form an unsubstituted or substituted heterocycle; and wherein each alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, alkylaryl, and alkylheteroaryl may be substituted or unsubstituted;
[0340] wherein the nitrogen in the benzoxazepine ring may optionally be a quaternary nitrogen; and all enantiomers, diastereomers, tautomers, pharmaceutically acceptable salts, hydrates, solvates, complexes, polymorphs, metabolites, and prodrugs thereof;
[0341] provided that, (i) when R is hydrogen at position 7 of the benzoxazepine ring, R2 is not hydrogen, alkyl, haloalkyl or alkoxyalkyl, (ii) when R3 is oxo, Ri is not oxo or -C(=0)NHR5; (iii) when R2 is H, Ri is not phenyl; and (iv) when R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsubstituted or substituted heterocycle, Ri is not oxo.
[0342] The invention further provides a number of more preferred structures that fall within the general structure of formula I-a-1. Preferred compounds of the present invention include:
[0343]• Compounds of formula I-a-1 wherein n, and R1-R4 are as in formula I-a-1 , and wherein each R is independently selected from the group consisting of Z, OCZ3, R5, OR5, CN, N02, N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, and -P(=0)R8R9, wherein R8 and R9 are
independently OR5, and wherein each R5 is independently hydrogen, or an unsubstituted or substituted alkyl, alkylaryl, aryl, or heterocyclyl.
[0344] • Compounds of formula I-a-1 wherein R is OR5 at position 7 of the benzoxazepine ring, n, and R1-R4 are as in formula I-a-1, and wherein R5 is selected from the group consisting of hydrogen, or an unsubstituted or substituted alkyl, alkylaryl, aryl, or
[0345]heterocyclyl. In one most preferred embodiment, R is methoxy.
[0346] • Compounds of formula I-a-1 wherein n, R, Rl s R3, and R4 are as in formula I-a-1, and wherein R2 is selected from the group consisting of (i) R5, (ii) -C(=0)R6, and (iii) -(CH2)mRio, wherein R5 is hydrogen, or an unsubstituted or substituted alkyl, aryl, alkylaryl, heterocyclyl or heteroaryl; wherein 5 is -NRi3Ri4, -C(=0)NRi3Ri4 or -(C=0)OR5; and wherein Ri3 and Ri4 together with the N to which they are bonded form an unsubstituted or substituted heterocycle; and wherein m is 1, 2, 3, 4, 5, or 6, and wherein R10 is R5 or
[0348] • Compounds of formula I-a-1 wherein n, R, Ri, R3, and R4 are as in formula I-a-1, and wherein R2 is selected from the group consisting of R5, -C(=0)(C=0)OR5,
[0349]-C(=0)NRi3Ri4, -CH2R10 and -C(=0)C(=0)NRi3Ri4, wherein R5 is hydrogen, or an unsubstituted or substituted alkyl, aryl, alkylaryl, heterocyclyl or heteroaryl; and wherein Ri3
[0351]and Ri4 are either each H or are bonded to make , wherein Rd is CH2, NH, O,
[0352]N-benzo[l,3]dioxo-5-yl, or N-C(=0)OC(R5)3, wherein the nitrogen in Rj may optionally be a quaternary nitrogen; and wherein Rio is R5 or (C=0)OR5. In one most preferred embodiment, R2 is -C(=0)C(=0)OH.
[0353] • Compounds of formula I-a-1 wherein n, R, Rls and R4 are as in formula I-a-1, and wherein R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsusbstituted or substituted heterocycle other than a piperazine.
[0354] • Compounds of formula I-a-1 wherein n, R, Ri and R2 are as in formula I-a-1, and wherein R3 and R4 together with the carbon atoms to which they are respectively attached, form an unsusbstituted or substituted cycloalkyl or heterocyclic ring.
[0355] Still other preferred compounds of the present invention include those of formula I-a-1, wherein
[0356] (a) n is 1 or 2, R is Z, OCZ3, R5, OR5, CN, N02, N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, or
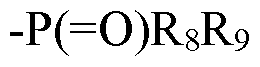 at position 7 or 8 of the benzoxazepine ring; or
(b) n is 1, R is Z, OCZ3, R5, OR5, CN, N02, -N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, or
at position 7 or 8 of the benzoxazepine ring; or
(b) n is 1, R is Z, OCZ3, R5, OR5, CN, N02, -N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, or
 at position 6 of the benzoxazepine ring; or
at position 6 of the benzoxazepine ring; or
[0357] (c) R2 is C(=0)R6, wherein R6 is selected from the group consisting of -C(=0)R5, -C(=0)OR5, -C(=0)NRi3Ri4, and (CH2)bNRi3Ri4, wherein b=0, and Ri3 and Ri4 are
[0359] either each H or are bonded to make ' , wherein Rd is O, CH2, or NRa; and Ra is H, alkoxy, C(=0)OC(CH3)3, or (Ci-C6 alkyl)-aryl, wherein the aryl is a disubstituted phenyl or a benzo[l,3]dioxo-5-yl group, and wherein the nitrogen in NRa may optionally be a quaternary nitrogen; or
[0360] (d) R2 is R5 or (CH2)mRi0, wherein Rio is selected from the group consisting of R5, -C(=0)N(R5)2, -(C=0)OR5, or -OR5; and m is 1, 2, 3, 4, 5, or 6.
[0361] More preferred compounds of (a) include Rg and R9 being independently OR5. Also in (a)-(d), more preferred compounds of (a)-(d) include each R5 being independently hydrogen, or an unsubstituted or substituted alkyl, alkylaryl, aryl, or heterocyclyl.
[0362] The preferred compounds of the invention specifically include those of formula I-a-1, wherein n is 1 and R is OR5, OCZ3, Z, CN, R5, N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, or
[0363]-P(=0)(OR5)2, N02 at position 6, 7 or 8 of the benzoxazepine ring; or n is 2, each R is independently OR5 at positions 7 and 8 of the benzoxazepine ring.
[0364] The more preferred compounds of the invention specifically include those of formula I-a-1, wherein:
[0365] A) n is 1 , R is OR5 or OCZ3 at position 7 of the benzoxazepine ring, and R2 is (i) hydrogen; (ii) R5, (iii) (CH2)mRi0, wherein m is 1, 2, 3, 4, 5, or 6, and wherein Rio is R5 or (C=0)OR5; (iv) -C(=0)C(=0)OR5; (v) -C(=0)NRi3Ri4 or (vi) -C(=0)C(=0)NRi3Ri4, wherein N Rd
[0366] Ri3 and Ri4 are either each H or are bonded to make \N ' / , wherein Rd is CH2, NH,
[0367]O, N-benzo[l,3]dioxo-5-yl, or N-C(=0)OC(R5)3, wherein the nitrogen in Rj may optionally be a quaternary nitrogen; or R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsubstituted or substituted heterocycle other than a piperazine; or
[0368] B) n is 1, R is Z, CN, R5, N(R5)2, -C(=0)N(R5)2, -C(=0)OR5, or -P(=0)(OR5)2 at position 7 of the benzoxazepine ring, and R2 is R5; or
C) n is 1 , R is N02 at position 8 of the benzoxazepine ring, and R2 is (i) hydrogen; (ii) R5, (iii) -C(=0)C(=0)OR5; or (iv) -C(=0)NRi3Ri4, wherein Ri3 and RM are either each H or N Rd
[0369]are bonded to make \ / , wherein Rd is CH2, NH, O, NC(=0)OC(R5)3, or
[0370] N-benzo[l,3]dioxo-5-yl, wherein the nitrogen in Rd may optionally be a quaternary nitrogen; or R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsubstituted or substituted heterocycle other than a piperazine; or
[0371] D) n is 2, each R is independently OR5 at positions 7 and 8 of the benzoxazepine ring, and R2 is (i) hydrogen; (ii) C(=0)C(=0)OR5; or (iii) -C(=0)NRi3Ri4, wherein Ri3 and RM are
[0373]either each H or are bonded to makeN ' , wherein Rj is CH2, NH, O,
[0374] N-benzo[l,3]dioxo-5-yl, or N-C(=0)OC(R5)3, wherein the nitrogen in Rj may optionally be a quaternary nitrogen; or
[0375] E) n is 1 , R is OR5 at position 6 of the benzoxazepine ring, and R2 and R3 together with the nitrogen and carbon to which they are respectively attached, form an unsubstituted or substituted heterocycle other than a piperazine; or
[0376] F) each of Rh R2, R3, and R4 is H, n=l, R is OR5, OCZ3, Z, CN, R5, N(R5)2,
[0377]-C(=0)N(R5)2, -C(=0)OR5, or -P(=0)(OR5)2, N02 and is at position 7 of the benzoxazepine ring.
[0378] The most preferred compounds of (A)-(F) include R being OR5 at position 7 of the benzoxazepine ring wherein each R5 is independently hydrogen, or an unsubstituted or substituted alkyl, alkylaryl, aryl, or heterocyclyl.
[0379] Still other preferred compounds are those represented by the structure of any one or more of formula I-b-1, 1-c-1, 1-d-1, 1-e-1, 1-f-1, 1-g-1, 1-h-1, and I-i- 1 , and their
[0380]pharmaceutically acceptable salts and hydrates.
[0381]
[0382] I-b-1
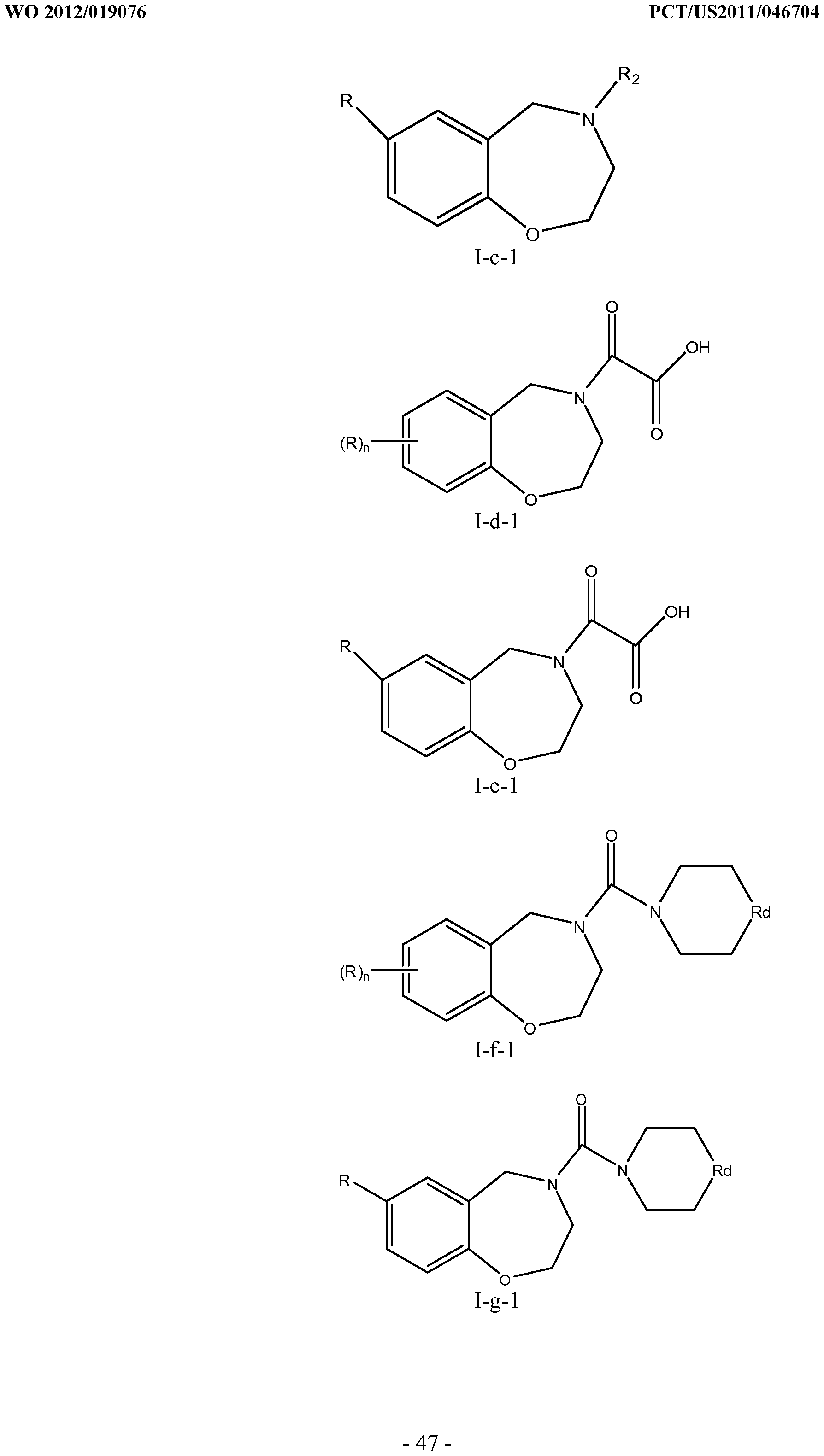
[0383]
[0384]
[0385]In these formulae, R, n and R2 are as in formula I-a-1 and Rd is CH2, NH, O,
[0386]N-benzo[l,3]dioxo-5-yl, or N-C(=0)OC(R5)3, wherein the nitrogen in Rj may optionally be a quaternary nitrogen.
[0387] The most preferred compounds of formula I-b-1 to I-i-1 include those where R is OR5 at position 7 of the benzoxazepine ring wherein each R5 is independently hydrogen, or an unsubstituted or substituted alkyl, alkylaryl, aryl, or heterocyclyl. Preferably, R is methoxy at position 7 of the benzothiazepine ring.
[0388] Examples of compounds that may be used in conjunction with the invention include, without limitation, SI, S2, S3, S4, S5, S6, S7, S9, SI 1, S12, SI 3, S14, SI 9, S20, S22, S23, S24, S25, S26, S27, S36, S37, S38, S40, S43, S44, S45, S46, S47, S48, S49, S50, S51, S52, S53, S54, S55, S56, S57, S58, S59, S60, S61, S62, S63, S64, S66, S67, S68, S69, S70, S71, S72, S73, S74, S75, S76, S77, S78, S79, S80, S81, S82, S83, S84, S85, S86, S87, S88, S89, S90, S91, S92, S93, S94, S95, S96, S97, S98, S99, S100, S101, S102, S103, S104, S107, S108, S109, S110, Si l l, S112, S113, S114, S115, S116, S117, S118, S119, S120, S121, S122, S123, S136, S137, S138, S139, S140, S146, S147, S148, S149, S150, S151, S152, S153, S156, S157, S159, S160, S161, S166, S167, S182, S186, S189, S203, S217, S251, S252, S258, S277, S279, S282, S291, S293, S296, S301, S302, S306, S311, S312, S313, S318, S322, S324, S326, S331, S335, S337, S351, S352, S353, S354, S397, S398, S399, S423, S454, S463, S466, S470, S473 and S477, as herein defined. In certain embodiments, the compounds are isolated and substantially pure.
[0389] The named "S" compounds described herein have the following structures:
[0390]
[0391]
[0392]-49-

[0393]
[0395]
[0397]
[0399]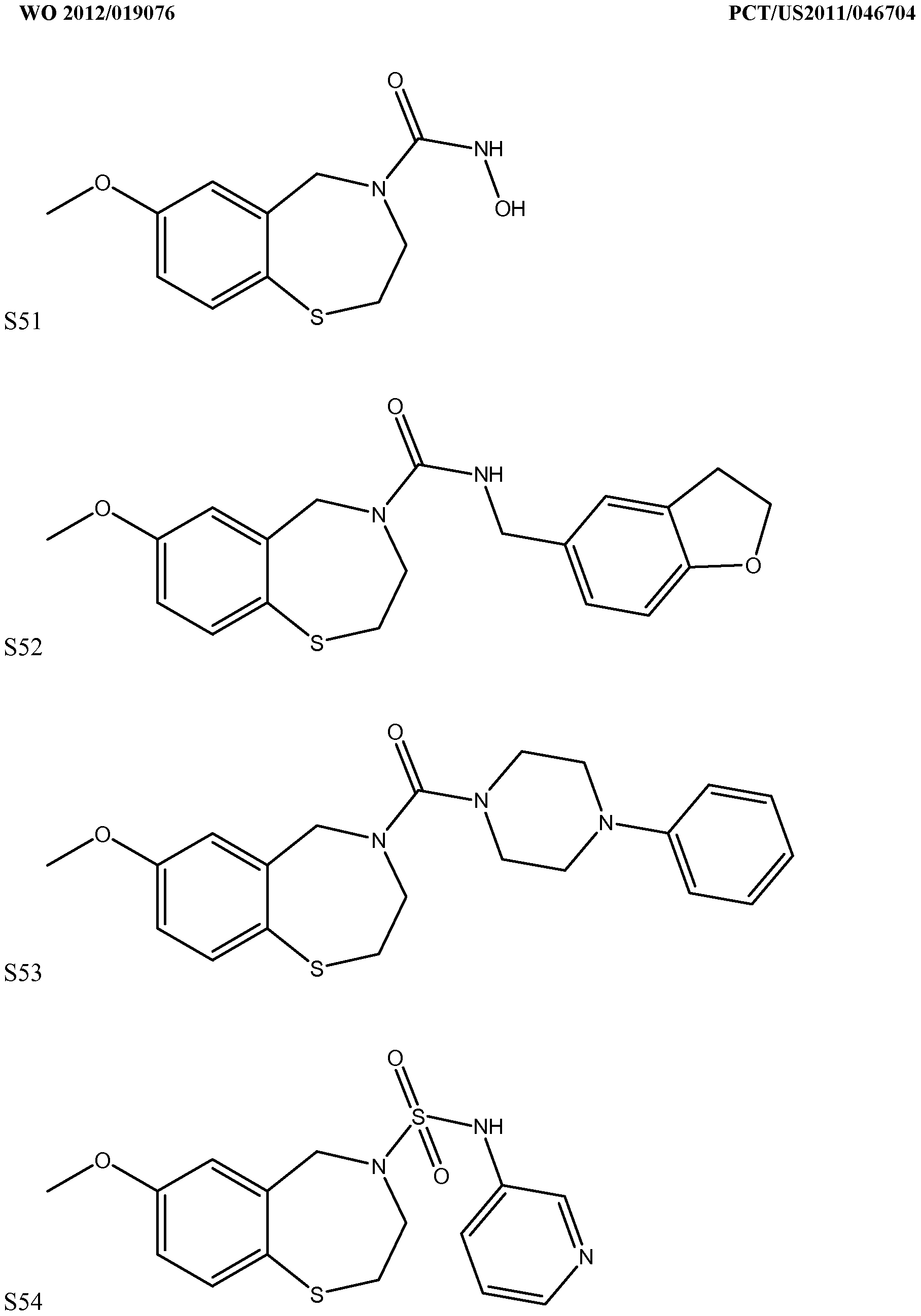
[0400]-57-

[0401]
[0403]
[0405]
[0407]
[0408]-62-


[0409]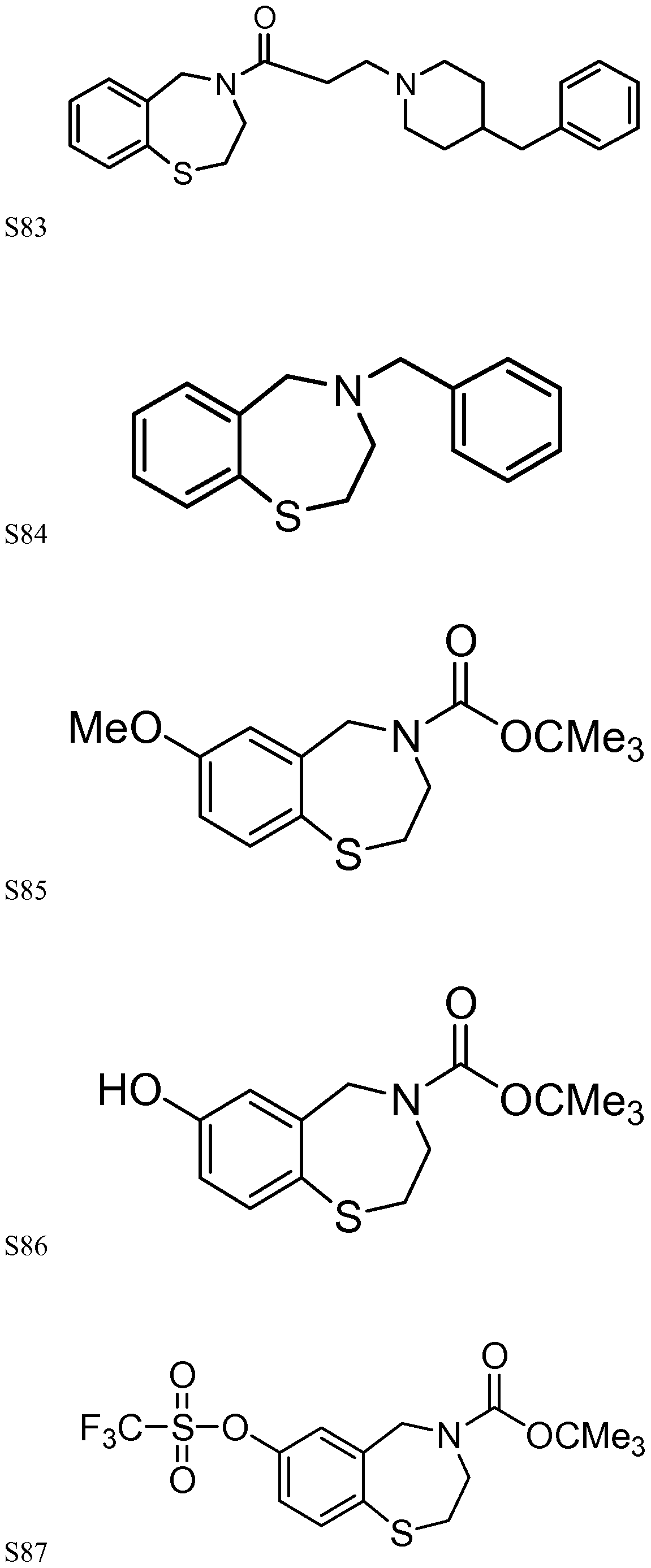
[0411]

[0412]S89

[0413]
[0415]
[0416]-67-


[0417]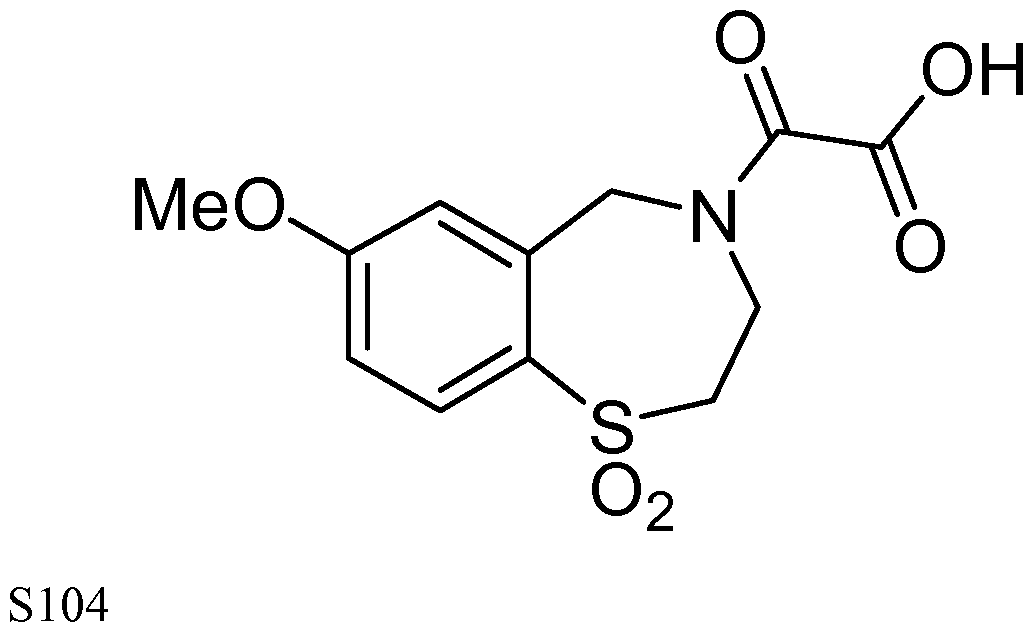

[0419]
[0421]
[0424]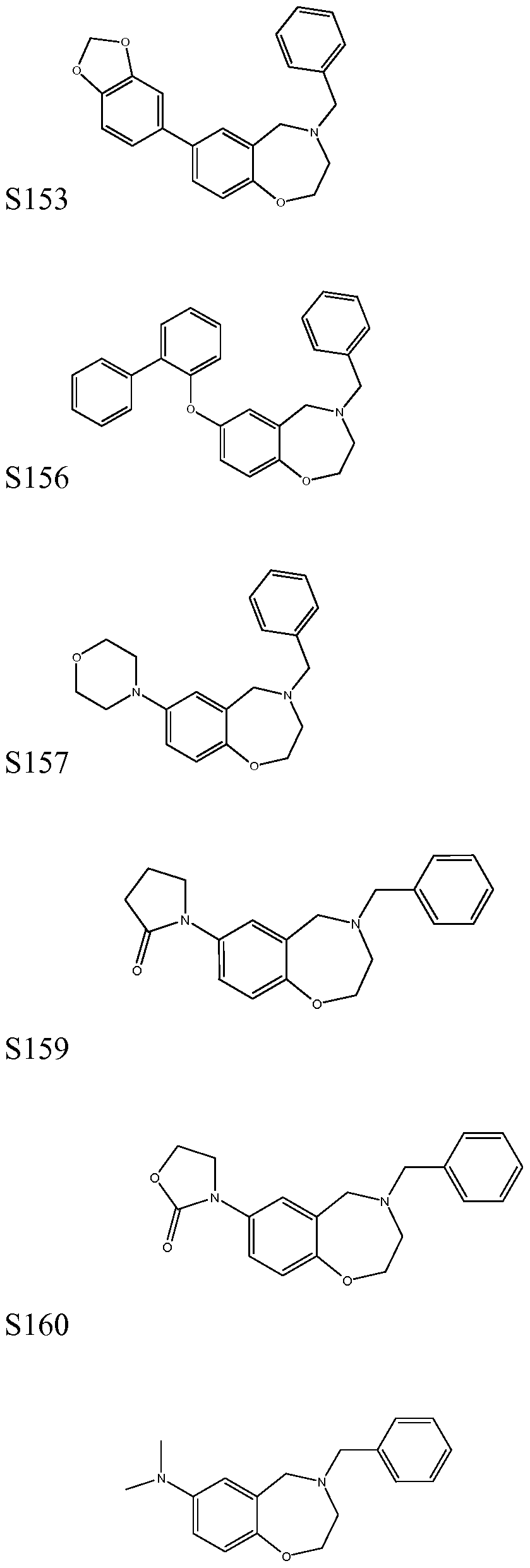
[0425]S161

[0427]
[0429]
[0430] S189

[0432]
[0433]S203

[0435]
[0436]S217

[0437]S251

[0438]S252

[0439]S258


[0440]

[0442]
[0443]S301
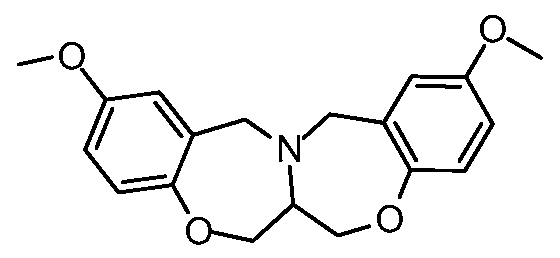
[0444]S302

[0445]S306

[0446]S311

[0448]
[0449]
[0450]
[0451] -80-
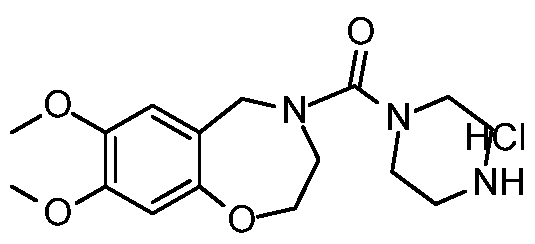
[0452]S335

[0453]S337

[0454]S351

[0455]S352
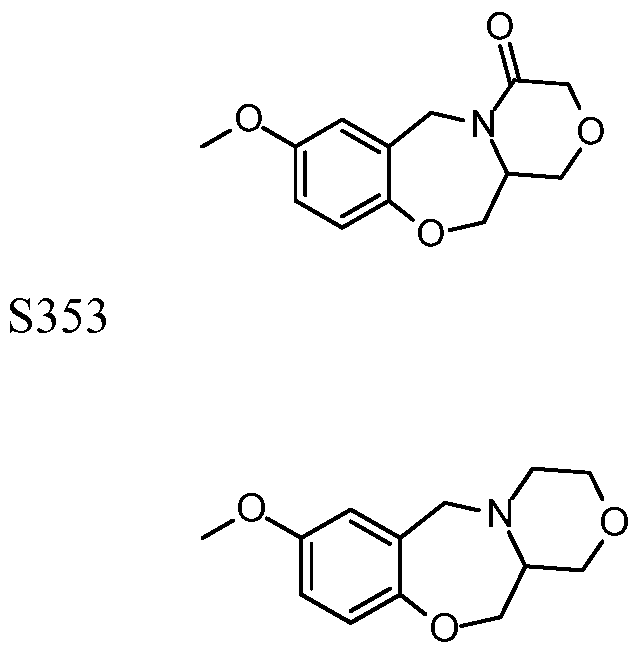
[0456] S354

[0457]S397

[0459]
[0461]
[0462]S423
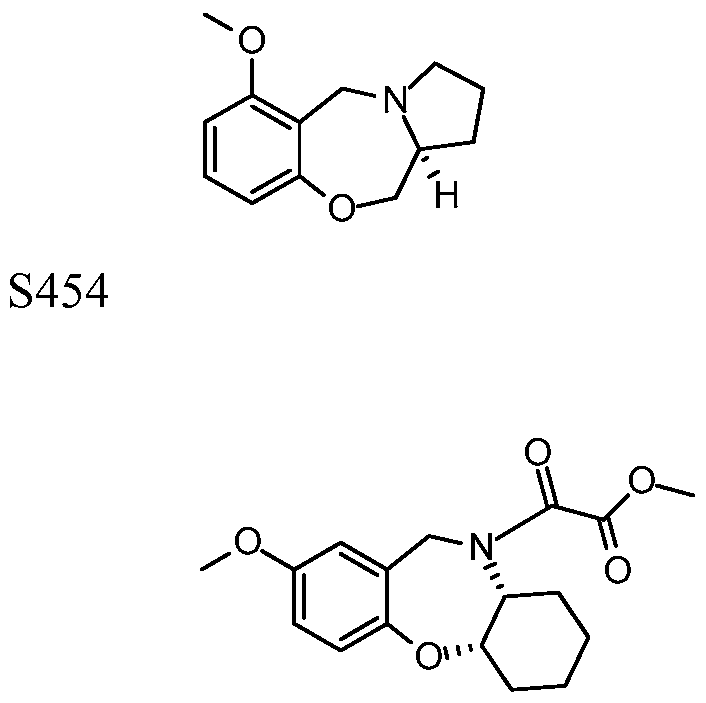
[0463] S463

[0465]
[0467]
[0469]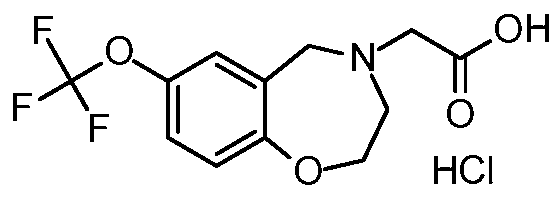
[0471]In one embodiment of the present invention, for compounds of Formula I, if R2 is C=0(R5) or SO2R7, then R is at positions 6, 7 or 9 on the benzothiazepine ring.
[0472] In another embodiment of the invention, for compounds of Formula I, if R2 is C=0(R5) or SO2R7, then each R is independently selected from the group consisting of H, halogen, -OH, -NH2, -NO2, -CN, -N3, -SO3H, acyl, alkyl, alkylamino, cycloalkyl, heterocyclyl, heterocyclylalkyl, alkenyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-) arylamino; wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, heterocyclyl, heterocyclylalkyl, alkenyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino may be substituted with one or more radicals independently selected from the group consisting of halogen, N, O, -S-, -CN, -N3, -SH, nitro, oxo, acyl, alkyl, alkoxyl, alkylamino, alkenyl, aryl, (hetero-)cycloalkyl, and (hetero-)cyclyl.
In another embodiment of the invention, for compounds of Formula I, if R2 is C=0(R5) or S02R7, then there are at least two R groups attached to the benzothiazepme ring.
[0473]Furthermore, there are at least two R groups attached to the benzothiazepme ring, and both R groups are attached at positions 6, 7, or 9 on the benzothiazepme ring. Still furthermore, each R is independently selected from the group consisting of H, halogen, -OH, -NH2, -N02, -CN, -N3, -SO3H, acyl, alkyl, alkylamino, cycloalkyl, heterocyclyl, heterocyclylalkyl, alkenyl, (hetero-)aryl, (hetero-)arylthio, and (hetero-)arylamino; wherein each acyl, alkyl, alkoxyl, alkylamino, cycloalkyl, heterocyclyl, heterocyclylalkyl, alkenyl, (hetero-)aryl,
[0474](hetero-)arylthio, and (hetero-)arylamino may be substituted with one or more radicals independently selected from the group consisting of halogen, N, O, -S-, -CN, -N3, -SH, nitro, oxo, acyl, alkyl, alkoxyl, alkylamino, alkenyl, aryl, (hetero-)cycloalkyl, and (hetero-)cyclyl.
[0475] In another embodiment of the invention, for compounds of Formula I, if R2 is
[0476]C=0(R5), then R5 is selected from the group consisting of -NR16, -(CH2)zNRi5Ri6, NHNHRie, NHOH, -ORis, CONH2NHRi6, CONRie, CH2X, acyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, aryl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl may be substituted with one or more radicals
[0477]independently selected from the group consisting of halogen, N, O, -S-, -CN, -N3, nitro, oxo, acyl, alkyl, alkoxyl, alkylamino, alkenyl, aryl, (hetero-)cycloalkyl, and (hetero-)cyclyl.
[0478] In another embodiment, the present invention provides use of the compounds of Formula II in the method of the invention. Formula II is
[0479] wherein R=OR', SR', NR, alkyl, or halide and R = alkyl, aryl, or H, and wherein R can be at position 6, 7, 8, or 9 of the benzothiazepme ring. Formula II is discussed also in co-pending application 10/680,988, the disclosure of which is incorporated herein in its entirety by reference.
wherein R=OR', SR', NR, alkyl, or halide and R = alkyl, aryl, or H, and wherein R can be at position 6, 7, 8, or 9 of the benzothiazepme ring. Formula II is discussed also in co-pending application 10/680,988, the disclosure of which is incorporated herein in its entirety by reference.
[0480] It is recognized in general that the -S- or -O- of the ring in all embodiments disclosed herein can instead be replaced by -CH2- or by -NH- with such compounds being expected to be useful by reducing calcium "leak" or calcium current through the RyR channel, stabilizing
gating of the RyR channel, or increasing the binding of calstabin to the RyR complex in the subject.
[0482] The compounds of the invention, such as the compounds of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1 , 1-b-1 , 1-c-1 , 1-d-1 , 1-e-1 , 1-f-1 , I-g-1 , 1-h-1 , 1-i-1 , or Formula II, reduce the open probability of RyRs and decrease the calcium current through such channels by increasing binding of calstabin (FKBP 12 or calstabinl , and FKBP12.6, also known as the cardiac calstabin2) binding affinity. Therefore, the compounds of the invention are useful for the treatment and/or prevention of disorders and conditions associated with abnormal function of RyRs, particularly RyRl and RyR2, where such disorders and conditions are characterized by an increase in the open probability of, and in increase in the calcium current through, RyR.
[0483] In accordance with the methods of the present invention, a "decrease" or "disorder" in the level of RyR-bound FKBP in cells of a subject refers to a detectable decrease, diminution or reduction in the level of RyR-bound FKBP in cells of the subject. Such a decrease is limited or prevented in cells of a subject when the decrease is in any way halted, hindered, impeded, obstructed or reduced by the administration of compounds of the invention, such that the level of RyR-bound FKBP in cells of the subject is higher than it would otherwise be in the absence of the administered compound.
[0484] The level of RyR-bound calstabin (FKBP) in a subject is detected by standard assays and techniques, including those readily determined from the known art (e.g., immunological techniques, hybridization analysis, immunoprecipitation, Western-blot analysis, fluorescence imaging techniques and/or radiation detection, etc.), as well as any assays and detection methods disclosed herein. For example, protein is isolated and purified from cells of a subject using standard methods known in the art, including, without limitation, extraction from the cells (e.g., with a detergent that solubilizes the protein) where necessary, followed by affinity purification on a column, chromatography (e.g., FTLC and HPLC), immunoprecipitation (with an antibody), and precipitation (e.g., with isopropanol and a reagent such as Trizol). Isolation and purification of the protein is followed by electrophoresis (e.g., on an SDS- polyacrylamide gel). A decrease in the level of RyR-bound FKBP in a subject, or the limiting or prevention thereof, is determined by comparing the amount of RyR-bound FKBP detected prior to the administration of a compound of Formula I, I-a', I-a, I-b, I-c, I-d, I-e, I-f, I-g, I-h, I-i, I-j, I-k, 1-1, 1-m, I-n, I-o, I-p, I-a-1 , 1-b-1 , 1-c-1 , 1-d-1 , 1-e-1 , I-f- 1 , I-g-1 , 1-h-1 , 1-i-1 , or
Formula II, (in accordance with methods described below) with the amount detected a suitable time after administration of the compound.
[0485] A decrease in the level of RyR-bound calstabin (FKBP) in cells of a subject is limited or prevented, for example, by inhibiting dissociation of FKBP and RyR in cells of the subject; by increasing binding between FKBP and RyR in cells of the subject; or by stabilizing the RyR-FKBP complex in cells of a subject. As used herein, the term "inhibiting dissociation" includes blocking, decreasing, inhibiting, limiting or preventing the physical dissociation or separation of an FKBP subunit from an RyR molecule in cells of the subject, and blocking, decreasing, inhibiting, limiting or preventing the physical dissociation or separation of an RyR molecule from an FKBP subunit in cells of the subject. As further used herein, the term "increasing binding" includes enhancing, increasing, or improving the ability of
[0486]phosphorylated RyR to associate physically with FKBP (e.g. , binding of approximately two fold or, approximately five fold, above the background binding of a negative control) in cells of the subject and enhancing, increasing or improving the ability of FKBP to associate physically with phosphorylated RyR (e.g., binding of approximately two fold, or,
[0487]approximately five fold, above the background binding of a negative control) in cells of the subject. Additionally, a decrease in the level of RyR-bound FKBP in cells of a subject is limited or prevented by directly decreasing the level of phosphorylated RyR in cells of the subject or by indirectly decreasing the level of phosphorylated RyR in the cells (e.g., by targeting an enzyme (such as PKA) or another endogenous molecule that regulates or modulates the functions or levels of phosphorylated RyR in the cells). In one embodiment, the level of phosphorylated RyR in the cells is decreased by at least 10% in the method of the present invention. In another embodiment, the level of phosphorylated RyR is decreased by at least 20%.
[0488] The efficacy of compounds S I -S I 07 in increasing binding of FKBP 12 or calstabin to RyRs, as shown by their EC50 values, can be found in published PCT application WO
[0489]07/024717 and U.S. patent application 1 1/506,285, the entire contents of which are hereby incorporated by reference. The EC50 data were obtained using an FKBP12.6 rebinding assay to determine the amount of FKBP12.6 binding to PKA-phosphorylated RyR2 at various concentrations (0.5 - 1000 nM) of these compounds. The EC50 values were calculated using Michaelis-Menten curve fitting. These previous studies have shown that some of the Rycal compounds have a higher biologic activity than JTV-519, a known modulator of RyR calcium-ion channels, as evidenced by their significantly lower EC50 values, as compared to that of JTV-519, which has an EC50 value of about 150nM.
In some preferred embodiments, the present invention provides methods and uses which comprise administering compounds of the invention having an EC50 value lower than 150 nM.
[0490] In some more preferred embodiments, the present invention provides methods and uses which comprise administering compounds of the invention having an EC50 value lower than 100 nM.
[0491] In some most preferred embodiments, the present invention provides methods and uses which comprise administering compounds of the invention having an EC50 value lower than 50 nM.
[0492]Methods of Synthesis
[0493] In general, the compounds of the present invention may be synthesized as described in published PCT application WO 07/024717 and U.S. patent application 11/506,285, the entire contents of which are hereby incorporated by reference.
[0494] In another embodiment, the present invention provides use of compounds of the following formula for preparing many of the compounds disclosed herein:
[0495]
[0497]wherein R=OR', SR', NR, alkyl, or halide and R = alkyl, aryl, or H, and wherein R can be at position 6, 7, 8, or 9 of the benzothiazepine ring. Various synthesis schemes are disclosed in application 12/263,435 the disclosure of which is expressly incorporated herein in its entirety by reference.
[0501] Fluo-4 AM, tetra-methyl rhodamine-ethyl ester (TMRM) and MitoSOX Red were from Molecular Probes/Invitrogen. Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), N-acetylcysteine (NAC), and laminin were from Sigma. All compounds were prepared as stock solutions in appropriate solvents. On the day of the experiment, stock
solutions were diluted to the desired final concentration in the bath solution; when required, the same concentration of solvent was added to the control solution. Benzyloxycarbonyl-Ile- Glu(OMe)-Thr-Asp(OMe)-Fluoromethylketone (Z-IETD-FMK; #:FK012), a preferential caspase-8, and Benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-Fluoromethylketone, and Benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-Fluoromethylketone (Z-DEVD- FMK; #:FK010), a preferential caspase-3 and -7 inhibitor, were obtained from MP
[0502]Biomedicals. In contrast to Z- and -FMK based inhibitors, the broad-spectrum caspase inhibitor Quinoline-carbonyl-Val-Asp-Difluorophenoxymethylketone (Q-VD-OPh; MP Biomedicals #:OPH109) has low toxicity, broad spectrum effects on caspases and no inhibitory effect on noncaspase enzymes. Na-Quinoline-2-carbonyl-LETD(OMe)- CH2OC6H3(2,6-F2) referred here as Q-LETD-OPh was custom-synthesized (95% purity, NeoMPs, Strasbourg, France). Q-LETD-OPh is a preferential caspase-8 inhibitor previously reported under the name TRP801. All inhibitors were dissolved at 10-2M in DMSO and stored at -20°C. Etanercept (Enbrel®, Pfizer France) was IP injected (2 mg/kg) 24h and lh before surgery.
[0503]Animal model and cell dissociation
[0504] Eight weeks-old Wistar Kyoto rats (Janvier, Le Genest-St-Isle, Fr) were used. The investigations conformed to the guidelines for the Care and Use of Laboratory Animals (NIH, N°. 85-23, revised 1996) and European directives (96/609/EEC). For ischemia/reperfusion protocol, rats were anesthetized with pentobarbital sodium [Abbott, North Chicago, IL; 40 mg/kg, intraperitonaly (i.p.)], and ventilated 60 times/min with a volume-cycled respirator. The left coronary artery was ligated at 1 to 2 mm from its origin with a 5-0 silk suture
[0505](Autosuture, Tyco Healthcare, Elancourt, France). After 15 min ligation, animals were randomly given vehicle (10% DMSO in 0.9% saline), Q-VD-OPh (i.p., 1 mg/kg in 10% DMSO) or QLETD-OPh (i.p., 1 mg/kg in 10% DMSO). After 30 min of occlusion, the ligation was removed and the left coronary artery reperfused. The chest cavity was compressed to remove any air before being hermetically sealed. Rats were randomly assigned to study groups according to the duration of left coronary reperfusion: from 1 hr (day 0-1 h) to 15 days and the caspase inhibitor treatment. Sham-operated animals were subjected to the same surgical procedure, but the ligation remained untied. In some experiments rats were pre- treated with S I 07 (25 mg/100 ml in the drinking water) one week prior to ischemia and up to 72 hr post-reperfusion). Similar procedure was used for WT and calstabin2 KO mice except for the duration of acute ischemia (1 hour).
Animals were sacrificed by rapid neck disarticulation and the heart was excised.
[0506]Single cardiomyocytes were enzymatically isolated from the left ventricles as described before 56. After being loaded with fluorescent indicators (see below), cardiomyocytes were plated on laminin-coated coverslips that made up the bottom of the perfusion chamber. Cells were superfused with standard Tyrode solution (mM): NaCl 117, KC1 5.7, NaHC03 4.4, KH2P04 1.5, MgCl2 1.7, HEPES 21, glucose 11, pH 7.4 adjusted with NaOH. Experiments were performed at room temperature (~24 °C). Cells were continuously stimulated at 1 Hz with 1-2 ms current pulses delivered via two platinum electrodes, one on each side of the perfusion chamber.
[0508] Thin sections (5 μιη) were prepared from the rat left ventricles, and stained with Haematoxylin- Eosin (HE) and Masson trichrome (MT) for light microscopy (x 10). The thickening of the myocardium was estimated on the HEstained heart sections, and the presence of collagen was analyzed on the MTstained ventricle sections. Results indicate the % of fibrosis area expressed as a percentage of collagen content area of myocardial tissue analyzed.
[0510] Cardiac function was assessed using echocardiography (Vivid 7, General Electric Healthcare) with a linear 14 MHz probe, 15 days after reperfusion. A two-dimensional view of the left ventricular (LV) was obtained at the level of the papillary muscles in a parasternal short-axis view. M-mode traces were recorded through the anterior and posterior walls.
[0511]Morpho-functionnal parameters were assessed using left ventricular end diastolic diameter (LVEDd), fractionnal shortening (FS) and E wave over A wave ratio (E/A) parameters.
[0512]Recording and analysis of ECG
[0513] Rats and mice were monitored with ECG recordings using a signal transmitter-receiver (RPC-1, DSI, St. Paul, MN) connected to a data acquisition system (IOX2, EMKA
[0514]Technologies, Paris, France). The data were collected continuously over 24 hr at a sampling rate of lOOOHz. Continuous digital recordings were analyzed off line using the software ECG- auto (ver 1.5.12.22, EMKA Technologies, Paris, France). ECG signals were digitally filtered between 0.1 and 1000 Hz and analyzed manually to detect arrhythmias over 2 hr of reperfusion in animal treated either with the vehicle (DMSO, i.p. injected), SI 07 (drinking water; 25 mg/lOOml) or Q-LETD-OPh (i.p. injected). The mean RR interval (ms) and the
standard deviation of all normal R intervals (SDNN, reflecting total autonomic variability) were calculated.
[0515]Cytosolic Ca2+ and Cell Shortening
[0516] Free cytosolic [Ca2+] was evaluated using the fluorescent Ca2+ indicator Fluo-4 and confocal microscopy in the line-scan mode using a Zeiss LSM 510 inverted confocal microscope (Zeiss, Le Pecq France) equipped with a 63X lens (H20 immersion, Numerical Aperture, NA=1.2). To enable comparisons between cells, changes in the Fluo-4 fluorescence signal (AF) were divided by the fluorescence immediately before a stimulation pulse given under control conditions (F0). The rising phase of Ca2+ transients was assessed by normalizing the peak amplitude over the time to peak (Amp/TT). Ca2+-sparks were analyzed as previously reported. Confocal images were obtained by line scanning along the long axis. Line-scan rate was set to 1.54 ms per line; 512 pixels x 3000 lines. Ca2+-spark frequency was calculated for each cardiomyocyte as the number of sparks recorded on 10 successive images collected at 3-4 different line locations. The SR Ca2+ content was measured in intact cardiomyocytes loaded with Fluo-4. After 5 min. of electrical field stimulation at 1 Hz, rapid perfusion of caffeine (20 mM) was used to release the SR Ca2+ store. Cell shortening was measured from the line-scan images and expressed as a percentage of the resting cell length.
[0517]Nitric oxide and mitochondrial ROS production.
[0518] DAF-FM was used to measure cytosolic NO level. Isolated cardiomyocytes were loaded with DAF-FM (2 μΜ) in Tyrode solution for 15 min at room temperature. Confocal images were obtained at 5 min intervals by measuring the emitted light at 515 nm after an excitation at 488 nm. MitoSOX Red was used to measure mitochondrial ROS production. Isolated cardiomyocytes were loaded with MitoSOX Red (5 μΜ) in Tyrode for 30 min at 37°C, followed by washout. Confocal images were obtained at 5 min intervals by measuring the emitted light at 585 nm after excitation at 488 nm. MitoSOX Red fluorescence was measured in five different areas in each cell and the signal was normalized to that at the start of the experiment. As a positive control, lmM of H202 was added at the end of each experiment and this resulted in a marked increase in the fluorescence signal in all cells (data not shown).
[0519]Mitochondrial Membrane Potential.
[0520] TMRM was used to measure mitochondrial membrane potential (ΔΨιη). Isolated cardiomyocytes were loaded with TMRM (10 nM) in Tyrode solution for 20 min at room
temperature, followed by washout in medium without TMRM. Confocal images of TMRM fluorescence were obtained by measuring the emitted light at 585 nm after excitation at 568 nm. To minimize the impact of subcellular variability in ΔΨιη, TMRM fiuorescence was measured in five different areas in each cell. Images were taken every 5 min and fluorescence signals were normalized to the fluorescence measured at the start of the experiment. At the end of each experiment, cells were exposed to the mitochondrial uncoupler FCCP (1 μΜ) to determine the dynamic range of the dye.
[0521]Sarcoplasmic Reticulum vesicle preparation.
[0522] About 200 mg of isolated rat left ventricular tissue was homogenized using a tissue mixer (Fisher Scientific) at the highest speed for 1 min with 2 volumes of: 20 mM Tris- maleate (pH 7.4), 1 mM EDTA and protease inhibitors (Roche). Homogenate was centrifuged at 4,000 g for 15 min at 4°C and the following supernatant was centrifuged at 40,000 g for 30 min at 4°C. The final pellet, containing the SR fractions, was resuspended and aliquoted using the following solution: 250 mM sucrose, 10 mM MOPS (pH 7.4), 1 mM EDTA and protease inhibitors. Samples were frozen in liquid nitrogen and stored at -80°C.
[0523]Immunoprecipitation and immunoblots analysis.
[0524] Cardiac SR microsomes (100 μg) were isotonically lysed in 1.0 ml of a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 20 mM NaF, 1.0 mM Na3V04, and protease inhibitors. An anti-RyR antibody (4 μg 5029 Ab) was used to immunoprecipitate RyR2 from 100 μg of cardiac SR microsomes. The samples were incubated with antibody in 0.5 ml of a modified RIPA buffer (50 mM Tris-HCl pH 7.4, 0.9% NaCl, 5.0 mM NaF, 1.0 mM Na3V04, 1% Triton- XI 00, and protease inhibitors) for 1 hr at 4°C. The immune complexes were incubated with protein A Sepharose beads (Sigma, St. Louis, MS) at 4°C for 1 hr and the beads were washed three times with buffer. Proteins were separated on SDS- PAGE gels (6% for RyR2, 15 % for calstabin) and transferred onto nitrocellulose membranes for 1 hr at 200 mA (SemiDry transfer blot, Bio-Rad). RyR2 S-nitrosylation was measured by developing immunoblots with both an RyR antibody (Affinity Bioreagents, Bolder, CO) and an anti-Cys-NO antibody (Sigma, St. Louis, MO) while RyR2 PKA phosphorylation at S2808 was measured by developing immunoblots with an anti-pS2808 antibody. The amount of calstabin2 that co-immunoprecipitated with RyR2 was determined from immunoblots developed with a calstabin antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Levels of RyR2 bound proteins were normalized to the total RyR2 immunoprecipitated (arbitrary units).
All immunoblots were developed and quantified using the Odyssey Infrared Imaging System (LICOR Biosystems, Lincoln, NE) and infrared-labeled secondary antibodies.
[0525]Single-channel Data Acquisition and Analysis
[0526] SR vesicles containing RyR2 were fused to planar lipid bilayers formed by painting a lipid mixture of phosphatidylethanolamine and phosphatidylcholine (Avanti Polar Lipids) in a 3: 1 ratio across a 200-um hole in polysulfonate cups (Warner Instruments) separating 2 chambers. The final concentration of lipids was 40 mg/ml dissolved in decane. Membrane thinning was assayed by applying a triangular wave test pulse. Typical capacitance values were 100 - 250 pF. The trans chamber (1.0 ml), representing the intra-SR (luminal) compartment, was connected to the head stage input of a bilayer voltage clamp amplifier. The cis chamber (1.0 ml), representing the cytoplasmic compartment, was held at virtual ground. The recording solutions consisted of: 1 mM EGTA, 250/125 mM Hepes/Tris, 0.64 mM CaCl2, pH 7.35 as cis solution and 53 mM Ca(OH)2, or 53 mM Ba(OH)2 when mentioned, and 250 mM Hepes, pH 7.35 as trans solution. The concentration of free Ca2+ in the cis chamber was calculated with WinMaxC program (version 2.50). SR vesicles were added to the cis side and fusion with the lipid bilayer was induced by making the cis side hyperosmotic by the addition of 400-500 mM CsCl. After the appearance of chloride channels, the cis side was perfused with the cis solution. Single-channel currents were recorded at 0 mV using a Bilayer Clamp BC-525D amplifier (Warner Instruments), and filtered with a low-pass Bessel filter eight pole (Warner Instruments) at 1 kHz and then sampled at 4 kHz. Data acquisition was performed by using Digidata 1322A and Axoscope 10 software (Axon Instruments). At the conclusion of each experiment, 5 μΜ ryanodine or 20 μΜ ruthenium red was applied to confirm RyR2 channel identity. The recordings were analyzed by using Clamp fit 10.1 (Molecular Devices) and Sigma Plot software (ver. 8.0, Systat Software).
[0528] The animals were sacrificed by a pentobarbital lethal injection. The heart was excised and perfused about 5 min by Langerdorf reverse way using a calcium-free washing solution (in mM: NaCl 117 mM, KC1 5.7, NaHC03 4.4, KH2P04 1.5, MgCl2 1.7, HEPES 21, glucose 11, taurine 20, pH 7.2 adjusted with NaOH). The left ventricle was than dissected. 2 mm3 was mechanical crushed in ice in the extraction buffer (HEPES lOmM pH 7.4, KC1 42mM, MgCl2 5mM, DTT lmM, CHAPS 0.5%, EDTA 0.1 mM supplemented with protease
inhibitors (PMSF ImM, leupeptin ^g/ml, pepstatin A ^g/ml, cytochalasin B ΙμΜ, chymopapain 10μg/ml, antipain ^g/ml)).
[0529] After centrifugation (10 min, 4°C, 2000g), 100 μg of supernatant was 1/10 diluted in a caspase activity buffer (Hepes 50mM pH 7.4, NaCl lOOmM, DTT lOmM, CHAPS 0.1%, EDTA ImM) to a final volume of 90 μΐ. The plate was incubated at 37°C during 3h in the dark with Ac-IETD-AMC. The measure was carried out by spectrofluorimetry
 380 nm;
380 nm;
[0530] 460 nm). Caspase activity was also assessed on isolated cardiomyocytes by measuring the activation of caspase-8 or caspase-3/7 by confocal imaging using a fluorescent caspase inhibitor (FLICA™, ImmunoChemistry Technologies LLC, Bloomington, MN). The carboxyfluorescein (FAM)-labeled caspase inhibitors FAM-IETD-FMK and FAM-DEVD- FMK were obtained from Abcys (Paris, France). Isolated cardiomyocytes were loaded following the commercial procedure and fluorescence was detected with excitation and emission wavelength at 488 nm and 515 nm, respectively.
[0532] Blood samples were withdrawn from the descending aorta at different time after reperfusion and allowed to clot 20 min at room temperature. Samples were then centrifugated at 2000 g during 10 min. at 4°C. Sera were aliquoted, quick frozen and stored at -20°C.
[0533]TNF-a levels were determined using xMAP luminex technology with a Biorad multiplex cytokine assay kit following the commercial procedure.
[0534]RNA extraction and real time RT-QPCR
[0535] Total RNA was extracted from approximately 15-25 mg of left ventricular cardiac tissue, using TRIzol reagent according to the manufacturer's protocol (Euromedex), then treated with DNase I (Invitrogen) at 37°C for 30 min. cDNA was synthesized using superscript II reverse transcriptase (Invitrogen) at 42°C for 50 min. RT-QPCR was performed in duplicates using a LightCycler rapid thermal cycler (Roche, France). Twenty-μΐ reaction mixture contained 10 μΐ of ABsolute QPCR SYBR Green Capillary Mix (Thermo Fisher Scientific, containing thermo start DNA polymerase, reaction buffer, deoxynucleoside trisphosphate mix, 3 mM MgCl2 and SYBR Green I dye), was added with 0.5 μΜ of appropriate primer mix, and 5 μΐ of cDNA. Forward and reverse primers for each gene were chosen on the basis of previously published sequences (ANF: forward primer
[0536]ATACAGTGCGGTGTCCAACA (SEQ ID NO: l) / reverse primer
[0537]CGAGAGCACCTCCATCTCTC (SEQ ID NO:2), fibronectin: forward primer
CCAGGCACTGACTACAAGAT (SEQ ID NO:3) / reverse primer
[0538]CATGATACCAGCAAGGAGT (SEQ ID NO:4), TNFR1 : forward primer
[0539]GGGATTCAGCTCCTGTCAAA (SEQ ID NO:5) / reverse primer
[0540]ATGAACTCCTTCCAGCTGGT (SEQ ID NO:6), TNFR2: forward primer
[0541]ATGGTGCCTCATCTGCC (SEQ ID NO:7) /reverse primer GGACCTGCTCATCCTTTG (SEQ ID NO: 8), caspase-8: forward primer CTGGGAAGGATCGACGATTA (SEQ ID NO:9) / reverse primer TGGTCACCTCATCCAAAACA (SEQ ID NO: 10), TNF-a: forward primer GTCGTAGCAAACCACCAAGC (SEQ ID NO: l 1) / reverse primer
[0542]TGTGGGTGAGGAGCACATAG (SEQ ID NO: 12)). The data were normalized to the levels of GAPDH.
[0543] The amplification program included the initial denaturation step at 95°C for 15 min, and 40 cycles of denaturation at 95°C for 1 s, annealing at 65°C for 10 s, and extension at 72°C for 20 s. Melting curves were used to determine the specificity of PCR products.
[0544]NFAT quantification
[0545] Proteins from 5 rats treated with DMSO, 6 rats treated with SI 07 and 4 rats treated with Q-LETD-OPh were extracted. The total protein concentration was determined with the Bio-Rad assay (Bio-Rad, Marnes-la-coquette, France). Equal amounts (50 μg) of cytosolic /nuclear protein were separated by 10% SDS-PAGE followed by immunob lotting with a Trans-Blot Semi-Dry (Bio-Rad), following the manufacturer's protocol. The following antibodies were used: rabbit GAPDH antibodies were from Abeam (1 :2500, Cambridge, United Kingdom) and rabbit NFAT4 antibody was from SantaCruz (1 :400). Western blots were revealed with the ECLplus reagent (GE Healthcare, Diegem, Belgique) and scanned with an Ettan-DIGE Imager (GE Healthcare). Densitometric analysis was performed with NIH Image/ImageJ, and the expression level of each protein was normalized to GAPDH for cytoplasmic signals. tBid quantification:
[0546] Heart tissues of Sham or I/R animals were homogenized directly into lysis buffer (10 mM Tris maleate pH6.8, 35 mM sodium fluorure, 1% triton, 1 mM sodium ortho vanadate, IX protease inhibitor). The lysates were centrifugated at 10,000 g for 5 minutes. Proteins were quantified with the DC Protein Assay (Biorad). 25 μg of proteins were loaded on 4-20% gradient gel of acrylamide. Proteins were transferred onto nitrocellulose membrane 0.2 μιη (GE Healthcare). The membrane were blocked (blocking buffer from Odyssey, LI-COR®
Biosciences) and then incubated with primary antibodies at room temperature for 1 hour: anti- BID (1 :400) (D-19: sc-6291, Santa Cruz Technology) and anti-GAPDH (1 : 1000) (FL-335: sc-25778, Santa Cruz Technology). The membrane was then incubated with secondary antibodies: anti-goat 680 nm (1 :30000) (BID) and anti-rabbit 800 nm (1 :30000) (GAPDH) for 45 minutes in the dark. After the final washes, the membrane was scanned using
[0547]ODYSSEY™ Infrared Imager (LI-COR® Biosciences).
[0549] Data are presented as mean±SEM. Statistical significance was assessed using the Student's t test (for paired or unpaired samples) or when three or more groups were compared, one-way analysis of variance (ANOVA) with a Newman-Keuls post hoc test. P < 0.05 was considered significant.
[0552] Figures 1A-G show effect of TNF-a and caspase-8 activation on RyR2 function in vitro. The following abbreviations are used: Q-LETD for caspase-8 inhibitor Q-LETD-OPh, Z-IETD for caspase-8 inhibitor Z-IETD-FMK, Z-DEVD for caspase-3/7 inhibitor Z-DEVD- FMK, Q-VD for broad spectrum caspase inhibitor Q-VD-OPh, and NAC for the antioxidant N-acetyl cysteine. In embodiment A are shown representative MitoSOX red fluorescence recorded at 0 min, 30 min and 60 min of TNF-a (10 ng/ml) or TNF-a+Q-LETD-OPh (10 μΜ) application in single ventricular rat cardiomyocytes. In embodiment B are shown mean data ± SEM of normalized MitoSOX red fluorescence after 60 min of TNF-a application. * indicates statistical difference compared to control conditions (P<0.05; n>20 cells in each conditions). Each caspases inhibitors (10 μΜ) were pre-incubated 15 min prior TNF-a application. For SI 07 experiments, the animals were orally treated with SI 07 (25 mg/100 ml, in drinking water) one week prior cells isolation. Note that caspase-8 inhibitors (Q-LETD-OPh and Z-IETD-FMK) and broad spectrum caspase inhibitor (Q-VD-OPh) prevents TNF-a induced mitochondrial ROS production whereas caspase-3/7 inhibitor (Z-DEVD-FMK) and SI 07 did not. In embodiment C are shown typical images of TNF-a-induced NO production measured with DAF-FM using confocal microscope. The right panel represents time change of normalized DAF-FM fluorescence in the presence of TNF-a (10 ng/ml) or TNF-a+ Q-LETD- OPh (10 μΜ). In embodiment D are shown mean data±SEM of normalized DAF-FM fluorescence after 60 min of TNF-a application. * indicates statistical difference compared to
control conditions (P < 0.05; n>20 cells in each conditions). Note that caspase-8 inhibitor (Q-LETD-OPh), broad spectrum caspase inhibitor (Q-VD-OPh) and eNOS inhibitor (L-NIO) prevent TNF-a induced NO production whereas ceramidase inhibitor (NOE) or SI 07 did not. In embodiment E are shown representative cardiac RyR2 immunoprecipitation and
[0553]immunoblots and bar graphs showing Cys nitrosylation of cardiac RyR2 and the amount of calstabin2 in the cardiac RyR2 complex. The bar graph shows the relative amount of calstabin2 associated with the RyR2 channel complex for each group determined by dividing the calstabin2 signals by the total amount of RyR2 that was immunoprecipitated (A.U.). The bar graph, depicting the relative amount of RyR2 S-nitrosylation for each group, was determined by dividing the Cys-NO signals by the total amount of RyR2 immunoprecipitated (A.U.). Data presented as mean±SEM. In embodiment F are shown representative RyR2 single-channel traces from control, TNF-a treated, and Q-LETD-OPh+TNF-a treated samples. RyR2 single channels were isolated from left ventricular cardiomyocytes treated 1 hr with TNF-a (10 ng/ml), or with Q-LETD-OPh (10 μΜ) followed by lh with TNF-a (10 ng/ml). Single channel activities were recorded at 150 nmol/L free cytosolic (cis) Ca2+ concentration and 53 mM Ca(OH)2 luminal (trans) at 0 mV. Channel openings are shown as upward deflections from the closed level (c-). Example of channel activity is shown at two different time scales (10 s for one upper trace and 1 s for two lower traces in each block) as indicated by dimension bars. The bar graph shows summary data of relative values of RyR2 Po of control, TNF-a treated and Q-LETD-OPh+TNF-a treated samples. *P<0.05 vs control. In embodiment G are shown spontaneous SR Ca2+ release events recorded in fluo-4-AM loaded intact cardiomyocytes by laser scanning confocal microscopy. Representative AF/F line scan images (1.54 ms/line) were recorded in absence (control) or after lh TNF-a incubation. Ca2+ sparks frequency is used as an index of diastolic SR Ca2+ leak. Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD), Z-IETD-FMK (Z-IETD), Z-DEVD-FMK (Z-DEVD), Q-VD-OPh (Q-VD). Data are expressed as mean ± SEM (*: p<0.05 vs. control; n> 30 cells in each conditions).
[0554] Figures 2A-I show roles of caspase-8 and RyR2 leak in myocardial reperfusion injury. In embodiment A are shown circulating levels of TNF-a after reperfusion compared to sham operated animals. Plasma level of TNF-a was maximal after 1 hr of reperfusion and returned to normal values within 6 hr of reperfusion. (*: p<0.05 vs. sham; n> 6 animal in each conditions). In embodiment B are shown normalized caspase-8 activity measured in left ventricular free wall homogenates after different time of reperfusion. Increased caspase-8 activity was maximal after 6h of reperfusion and return to normal values within the first 24 hr
of reperfusion. (n = 6 animals). In embodiment C are shown representative cardiac RyR2 immunoprecipitation and immunoblots and bar graphs showing Cys nitrosylation of cardiac RyR2 and depletion of calstabin2 from the cardiac RyR2 complex, 24 hr post-reperfusion in a rat model of ischemia-reperfusion (I/R) treated either with the vehicle (DMSO, i.p. injected), S107 (drinking water; 25 mg/lOOml) or Q-LETD-OPh (i.p. injected 15 min prior reperfusion). The relative amount of calstabin2 associated with the channel complex was determined by dividing the calstabin2 signals by the total amount of RyR2 immunoprecipitated (A.U.). The relative amount of RyR2 S-nitrosylation for each group was determined by dividing the Cys- NO signals by the total amount of RyR2 immunoprecipitated (A.U.). Data presented as mean ± S.E.M. In embodiment D are shown representative sections of TTC-stained hearts.
[0555]Quantification was done by normalizing the infarct area (IA) to the area at risk (AAR).
[0556]Treatment with Q-LETD-OPh or SI 07 reduces infarct size after 24 hr reperfusion. In embodiment E are shown ECG recorded in vivo after I/R. Typical ECGs acquired by telemetry over 2 hr of reperfusion in animal treated either with the vehicle (DMSO, i.p.
[0557]injected), SI 07 (drinking water; 25 mg/lOOml) or Q-LETD-OPh (i.p. injected 15 min prior reperfusion). Note the increase heart rate in DMSO injected animal. In embodiment F are shown typical ventricular ectopic beat (indicated by the arrows) recorded during the 2 hr of reperfusion in DMSO injected animal. In embodiments G-I are shown ECG analysis which provides various functional parameters, including cardiac frequency given by the RR interval (embodiment G), heart rate variability expressed as SDNN (embodiment H), and ventricular ectopic beat (VEB), also known as premature ventricular contraction (PVC), counts
[0558](embodiment I). (*: p<0.05 vs. sham; n> 6 animals in each conditions). Caspase-8 inhibitor, Q-LETD-OPh, is indicated as Q-LETD.
[0559] Figures 3A-C show left ventricular remodeling 15 days after reperfusion. In embodiment A are shown heart sections stained with Masson trichrome, which revealed a major increase in fibrosis that was prevented when animals where i.p. injected in Q-LETD- OPh, 15 min. prior to the reperfusion or when pre-treated with SI 07 (one week prior to ischemia and up to 72 hr post-reperfusion) compared to vehicle treated animals. (*: p<0.05 vs. sham; n> 6 animals in each conditions). Embodiment B shows that heart weight to body weight ratio was significantly increased in DMSO treated animals and unchanged in Q-LETD- OPh- or S107-treated animals (*: p<0.05 vs. sham; n> 10 animals in each conditions). In embodiment C are shown echocardiographic parameters analyzed 15 days after reperfusion. Left ventricular telediastolic diameter (LVtd, left panel), fractional shortening (FS, middle panel) and E wave over A wave ratio (E/A; right panel) are significantly affected in DMSO
treated animal. Echocardiography parameters were significantly enhanced when animals where i.p. injected in Q-LETD-OPh or with SI 07. (*: p<0.05 vs. sham; n> 8 animals in each conditions). Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD) and Q-VD- OPh (Q-VD).
[0560] Figures 4A-E show the diastolic SR Ca2+ leak via RyR2 channels after
[0561]ischemia/reperfusion contributes to the cardiac remodeling process. In embodiment A are shown representative cardiac RyR2 immunoprecipitation and immunoblots and bar graphs showing Cys nitrosylation and depletion of calstabin2 from the cardiac RyR2 complex, 2 weeks post-reperfusion. Levels of proteins in the RyR2 complex were normalized to the total amount of RyR2 (A.U.). Data presented as mean ± S.E.M. In embodiment B are shown representative RyR2 single-channel traces recorded at 150 nmol/L free cytosolic (cis) Ca2+ concentration and 53 mM Ba(OH)2 luminal (trans) at 0 mV. RyR2 channels were isolated from hearts 15 days post-reperfusion. Channel openings are shown as upward deflections from the closed level (c-). Example of channel activity is shown at two different time scales (10 s for one upper trace and 1 s for two lower traces in each block) as indicated by dimension bars. Summary data of relative values of RyR2 normalized Po under different treatment conditions are indicated in the labeled legend. The single channel Po at 150 nmol/L free cytosolic Ca2+ concentration was normalized to the Po at 5000 nmol/L free cytosolic Ca2+ concentration. *P<0.05 vs sham. # <0.05 vs DMSO. Data are the means ± S.E. of 5 to 9 experiments for each group. In embodiment C are shown western-blot showing the presence of NFAT4 in cytosolic and nuclear fractions from different cardiac samples. The low level of GAPDH in the nuclear fraction indicates that these fractions were not contaminated by cytosol. The histograms represent the ratio of nuclear NFAT4 to cytosolic plus nuclear NFAT4. In embodiment D are shown mRNA expression level of Atrial Natriuretic Factor (ANF) and fibronectin in left ventricular free wall 15 days after reperfusion. (*: p<0.05 vs. sham; n> 8 animals in each conditions). In embodiment E are shown mRNA expression level of TNF-a signaling cascade key proteins such as TNF-a receptor 1 and 2 (TNFR1 and TNFR2), caspase-8 (C8) and TNF-a. Note that ip injection 15 min prior reperfusion of Q- LETD-OPh or Q-VD-OPh or a pre -treatment with SI 07 (one week prior to ischemia and up to 72 hr post-reperfusion) normalized mRNA expression level compared to DMSO treated animals. (*: p<0.05 vs. sham; n> 8 animals in each conditions). Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD) and Q-VD-OPh (Q-VD).
[0562] Figures 5A-C show effect of TNF-a and caspase-8 activation on Ca2+ sparks frequency in cardiomyocytes. In embodiment A caspase-like activities were assessed using
the fluorescent caspase irreversible inhibitors FAM-DEVD-FMK (a preferential caspase-3/7 probe) or FAM-IETD-FMK (a preferential caspase-8 inhibitor). Specificity of each fluorescent probe was tested using the non- fluorescent inhibitor (i.e., Z-DEVD-FMK or Z-IETD-FMK, respectively; 10 μΜ) as well as the broad caspase inhibitor Q-VD-OPh (10 μΜ) in the presence of TNF-a (10 ng/ml). TNF-a significantly increased caspase-3-like and caspase-8-like activities. Caspase-8 inhibitors (Z-IETD-FMK and Q-LETD-OPh) prevented increased fluorescence of both FAM-based probes whereas Z-DEVD-FMK did not affect increase of FAM-IETD-FMK fluorescence. In embodiment B are shown TNF-a dissipated ΔΨιη in ventricular cardiomyocytes. The normalized TMRM fluorescence recorded under control conditions, during 60 min with 10 ng/ml TNF-a with or without the different caspases inhibitors (10 μΜ) or in cardiomyocytes isolated from animal treated in drinking water with SI 07 (25 mg/lOOml). Caspase-8 inhibitors (Q-LETD-OPh or Z-IETD-FMK) prevented ΔΨιη dissipation as well as the broad caspase inhibitor, Q-VD-OPh. Caspase-3-like inhibitor (Z-DEVD-FMK) or SI 07 did not prevent TNF-a-induced ΔΨιη dissipation. In embodiment C are shown Ca2+ sparks frequency recorded with or without TNF-a (lOng/ml) in presence of Q-LETD-OPh (ΙμΜ and 10μΜ) or in cardiomyocytes isolated from animal treated with SI 07 (25 mg/ 100ml of drinking water). None of the treatment affects sparks frequency in basal conditions compared to control. Similarly, combined Q-LETD-OPh and SI 07 treatment did not have additive effects on basal sparks frequency or in the inhibition level of TNF-a increased Ca2+ sparks frequency. Data are mean ± SEM. (*: p<0.05 vs. control; n> 15 cells in each conditions). Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD), Z-IETD-FMK (Z-IETD), Z-DEVD-FMK (Z-DEVD), Q-VD-OPh (Q-VD). Data are expressed as mean ± SEM. (*: p<0.05 vs. control; n> 20 cells in each conditions).
[0563] Figures 6A-E show effects of acute TNF-a incubation (10 ng/ml; lh) on Ca2+ transients recorded in fluo-4 AM-loaded intact cardiomyocytes by laser scanning confocal microscopy, in the presence of different caspases inhibitors, the anti-oxidant (NAC) or SI 07. In embodiment A are shown representative AF/F recorded in control, TNF-a, TNF-a + Q-LETD-OPh or TNF-a + Z-DEVD-FMK incubated cardiomyocytes. In embodiments B-D are shown average properties of Ca2+ transients such as amplitude (embodiment B), rate of Ca2+ transients rate of rise (Amp/TT) (embodiment C), and SR Ca2+ load (embodiment D) and fractional shortening (embodiment E), which are expressed as mean ± SEM. (*: p<0.05 vs. control; n> 20 cells in each conditions). Note that TNF-a triggered frequent arrhythmogenic Ca2+ transients that caspase-3 inhibitor (Z-DEVD-FMK, 10 μΜ) did not abolished compared
to Q-LETD-OPh treated cardiomyocytes (10 μΜ). Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD), Z-IETD-FMK (Z-IETD), Z-DEVD-FMK (Z-DEVD), and Q-VD-OPh (Q-VD).
[0564] Figure 7 shows Bid cleavage assessed by Western blot analysis in sham and IR hearts after 1 , 6 and 24 hours of reperfusion. tBid content was normalized to GAPDH and expressed as a percentage of the values obtained in sham for each time point. Values are expressed as mean±SEM (n=5-6 heart for each condition, *: p<0.05).
[0565] Figure 8 show representative sections (Top) of TTC-stained hearts. Quantification was done by normalizing the infarct area (I A) to the area at risk (AAR) (Bottom). Treatment with Q-LETD-OPh, SI 07 or etanercept reduces infarct size after 24 hr reperfusion.
[0566] Figures 9A-B show effect of ischemia reperfusion on calstabin2 KO mice. In embodiment A are shown representative sections (Top) of TTC-stained hearts. Quantification was done by normalizing the infarct area (I A) to the area at risk (AAR) (Bottom). Myocardial infarct size was significantly increased in calstabin2KO mice. Treatment with SI 07 reduces infarct size after 24 hr reperfusion in WT but not in calstabin2KO mice. In embodiment B are shown representative cardiac RyR2 immunoprecipitation and immunoblots and bar graphs showing amount of calstabin2 associated with RyR2, Cys-nitrosylation and PKA
[0567]phosphorylation level at S2808. Equivalent amounts of RyR2 were immunoprecipitated from SR cardiac microsomes using an anti-RyR2 antibody. The bar graph, depicting the relative amount of calstabin2 associated with the channel complex for each group, was determined by dividing the calstabin2 signals by the total amount of RyR2 immunoprecipitated (A.U.). The bar graph, depicting the relative amount of RyR2 S-nitrosylation and RyR2 PKA- phosphorylated at S2808 for each group, was determined by dividing the Cys-NO signals and pS2808 signals by the total amount of RyR2 immunoprecipitated (A.U.). Values are expressed as mean ± SEM (n>3 for each condition, *: p<0.05).
[0568] Figures lOA-C show effects of TNF-a in WT mice in vivo. In embodiment A are shown premature ventricular contractions count over 120 minutes after injection (left panel). Representative electrocardiogram traces recorded in mice injected with vehicle, TNF-a or TNF-a + Q-LETD-OPh (Bottom). In embodiment B, Bid cleavage was assessed by Western blot analysis in vehicle, TNF-a or TNF-a + Q-LETD hearts after 6 hours of I.V. injection. tBid content was normalized to total amount of Bid. In embodiment C are shown
[0569]representative cardiac RyR2 immunoprecipitation and immunoblots and bar graphs showing amount of calstabin2 associated with RyR2, Cys-nitrosylation and PKA phosphorylation level at S2808. Equivalent amounts of RyR2 were immunoprecipitated from SR cardiac
microsomes using an anti-RyR2 antibody. The bar graph, depicting the relative amount of calstabin2 associated with the channel complex for each group, was determined by dividing the calstabin2 signals by the total amount of RyR2 immunoprecipitated (A.U.). The bar graph, depicting the relative amount of RyR2 S-nitrosylation and RyR2 PKA-phosphorylated at S2808 for each group, was determined by dividing the Cys-NO signals and pS2808 signals by the total amount of RyR2 immunoprecipitated (A.U.). Values are expressed as mean ± SEM (n>6 for each condition, *: p<0.05).
[0570] Figures 11A-D show Ca2+ transients recorded in fiuo-4 AM-loaded intact
[0571]cardiomyocytes by laser scanning confocal microscopy after 15 days of reperfusion in an in vivo model of ischemia-reperfusion. Animals were treated with DMSO (vehicle) Q-LETD- OPh, Q-VD-OPh, or SI 07. In embodiments A-D are shown average properties of Ca2+ transients such as amplitude (embodiment A), rate of Ca2+ transients rise (Amp/TT)
[0572](embodiment B), SR Ca2+ load (embodiment C) and fractional shortening (embodiment D) are expressed as mean ± SEM. (*: p<0.05 vs. control; n> 20 cells in each conditions). Caspases inhibitors are indicated as follow: Q-LETD-OPh (Q-LETD) and Q-VD-OPh (Q-VD).
[0573] Figure 12 shows representative cardiac RyR2 immunoprecipitation and immunoblots showing the level of PKA phosphorylation level at S2808. Equivalent amounts of RyR2 were immunoprecipitated from SR cardiac microsomes using an anti-RyR2 antibody.
[0574]Investigating Effect of TNF-q and Caspase-8 Activation on RvR2 Function in vitro
[0575] Acute application of TNF-a (lhr, 10 ng/ml) to freshly isolated control cardiomyocytes induced caspase-8 like and caspase-3 like activities sequentially (Fig. 5A-B). In addition, application of TNF-a resulted in a progressive and significant increase in the Mitosox Red fluorescence within 1 hr in control cells or in myocytes pre-incubated with a preferential caspase-3/7 inhibitor (Z-DEVD-FMK; 10μΜ) (103±10%, n=30 and 90.5±12%, n=29, respectively), whereas there was no change in fluorescence in cells pre-incubated either with preferential caspase-8 inhibitors (Q-LETD-OPh 10 μΜ or Z-IETD-FMK, 10 μΜ) or with broad spectrum caspase inhibitor (Q-VD-OPh, 10 μΜ) (Fig. 1A-B). Of note, this
[0576]mitochondrial ROS production was associated with a significant ΔΨιη depolarization which was inhibited by caspase-8 inhibitors (Fig. 5C-D). Several studies have reported that TNF-a increases NO production either acutely through eNOS activation or after induction of iNOS expression. Thus, the inventors tested the effect of TNF-a on NO production using DAF-FM which increases its fluorescence when oxidized by NO. TNF-a application caused a progressive increase in the DAF-FM fluorescence which was inhibited by caspase-8 inhibitors
(32 ± 5%, n=20 vs 0.2 ± 0.8% n=20; P <0.05, Fig. 1C-D). Thus, TNF-a induced NO production seems to require caspase-8 activation. Under these pro-inflammatory conditions, simultaneous production of superoxide anion (02~) and NO can generate peroxynitrite formation. Among proteins involved in excitation-contraction coupling, RyR2 is highly sensitive to peroxynitrite and subsequent S-nitrosylation. In the present study, acute incubation of cardiomyocytes with TNF-a (10 ng/ml) for 1 hr was sufficient to cause RyR2 S-nitrosylation and calstabin2 depletion from RyR2 complexes (Fig. IE). This was accompanied by an increase in open probability (Po) of RyR2 channels incorporated into planar lipid bilayers (Fig. IF) and an increase in Ca2+-spark frequency in intact ventricular cardiomyocytes (Fig. 1G). In the presence of the caspase 8 inhibitor, QLETD-OPh, the TNF-a induced changes in RyR2 S-nitrosylation, calstabin2 binding to the RyR2 complex, RyR2 Po and Ca2+-sparks frequency were prevented (Fig. IE, F, G). The increase in Ca2+ sparks frequency was also prevented by the RyR Ca2+ release channel stabilizer "rycal" SI 07 (Fig. 1G). However, SI 07 treatment did not prevent mitochondrial inner membrane depolarization (ΔΨιη) (Fig. 5D), mitochondrial ROS production (Fig. 1A-B), or NO production (Fig. 1C, D). The antioxidant N-acetyl cysteine (NAC) also normalized Ca2+-spark frequency (Fig. 1G). Moreover, TNF-a decreased the Ca2+ transient amplitude, Ca2+ release kinetics, SR Ca2+ load and cell shortening (Fig. 6A-E). Q-LETD-OPh prevented the TNF-a induced decrease in the Ca2+ transient amplitude, Ca2+ release kinetics, SR Ca2+ load and cell shortening (Fig. 6A-E). When experiments were performed in presence of Q-LETD-OPh, QV-D-OPh, SI 07 or NAC, Ca2+ transients as well as cell shortening and SR Ca2+ load were increased in presence of TNF-a compare to control condition (Fig. 6B-E). Thus, TNF-a mediated ROS/NO production via caspase-8 activation, increased RyR2 S-nitrosylation and SR Ca2+ leak.
[0577]Roles of Caspase-8 and RyR2 Leak in Myocardial Reperfusion Injury
[0578] In order to determine whether the TNF-a induced SR Ca2+ leak via RyR2 channels contributes to reperfusion injury, the inventors performed 30 min. of ischemia followed by reperfusion in vivo in rats. TNF-a plasma levels were detected at 1 hr and returned to baseline 6 hr post-reperfusion (276 ± 48 pg/ml at 1 hr, n=6; Fig. 2A). In parallel, cardiac caspase-8 activity was also significantly increased by 1 hr following reperfusion, peaked at 6 hrs and returned to baseline by 24 hr (Fig. 2B). RyR2 S-nitrosylation and calstabin2 depletion were also observed 24 hr post-reperfusion (Fig. 2C). SI 07 (25 mg/100 ml, in drinking water) treatment for one week prior to surgery prevented calstabin2 depletion from the RyR2 complex but did not affect S-nitrosylation of the channel (Fig. 2C). In contrast, Q-LETD-OPh
treatment (1 mg/kg ip) 15 min prior to reperfusion, inhibited both RyR2 S-nitrosylation and depletion of calstabin2 from the RyR2 complex (Fig. 2C). In addition, both SI 07 and
[0579]Q-LETD-OPh significantly reduced myocardial infarct size [Infarct Area/ Area At Risk (IA/AAR)] compared to DMSO-treated rats (Fig. 2D). The severity of the ischemic insult was similar in the different groups, as shown by the ratios of AAR relative to ventricle area (V) (AAR V= 39 ± 8% in DMSO treated, n=10; 35 ± 6% in Q-LETD-OPh treated, n=8; 38 ± 6% in SI 07 treated, n=8). SR Ca2+ leak is thought to play a role in triggering arrhythmias during the early phase of reperfusion. The inventors observed numerous ventricular extrasystoles and sustained ventricular tachycardia during the first 12 hrs of reperfusion (Fig. 2E-I). QLETD- OPh and SI 07 treatment both significantly reduced arrhythmias (Fig. 2E, G, H, I). Thus, inhibition of caspase-8 and prevention of calstabin2 depletion from the RyR2 complex with SI 07 prevents early reperfusion injury and associated arrhythmias.
[0580]Role of Caspase-8 and RvR2 Leak in Left Ventricular Remodeling
[0581] The longer-term effects of either early caspase-8 inhibition or SI 07 treatment on left ventricular remodeling 15 days after myocardial reperfusion were subsequently analyzed and compared. Histological analyses of the left ventricle were performed using Masson trichrome staining to detect collagen fibers. Fifteen days after myocardial reperfusion, there was an increase in extracellular matrix (i.e. fibrosis) that was prevented when animals were treated with Q-LETD-OPh (1 mg/kg ip), 15 min. prior to the reperfusion or pre-treated with SI 07 (25 mg/100 ml in the drinking water) one week prior to ischemia and up to 72 hr post-reperfusion, compared to vehicle treated animals (Fig. 3 A). As an index of hypertrophy, HW/BW ratio was significantly increased in DMSO treated animals and unchanged in Q-LETD-OPh or S107-treated animals (Fig. 3B). Following ischemia/reperfusion the rats exhibited evidence of cardiac dysfunction with an increase in LVtd and a decrease in fractional shortening (Fig. 3C), both of which were significantly improved in animals treated with Q-LETD-OPh or SI 07 (Fig. 3C).
[0582] The cardiac ventricular remodeling observed 2 weeks post-reperfusion was associated with RyR2 S-nitrosylation, calstabin2 depletion from the RyR2 complex (Fig. 4A), and an increase in RyR2 channel open probability measured under conditions corresponding to diastole with low activating [Ca2+] (150 nM) (Fig. 4B), consistent with a diastolic SR Ca2+ leak. Once again, SI 07 inhibited depletion of calstabin2 from the RyR2 complexes without affecting S-nitrosylation of RyR2, whereas Q-LETD-OPh normalized both. RyR2 channel open probability, measured at 150 nM cytosolic Ca2+, was partially or totally reduced to that
observed in control channels from animals treated with SI 07 or Q-LETD-OPh, respectively. At the cellular level, Ca2+ transient amplitudes were decreased by about 20% and the rising phases as well as the decay time constants were significantly slower in vehicle treated animals (Fig. 7A). These changes were accompanied by decreased SR Ca2+ content and fractional cell shortening (Fig. 7B-D). Thus, SI 07 or Q-LETD-OPh treatment prevented altered Ca2+ handling and impaired cell shortening. Cytosolic Ca2+ regulates the nuclear translocation of some transcription factors and the expression of Ca2+ dependent genes known to contribute to ventricular remodeling. To confirm that the diastolic SR Ca2+ leak via RyR2 channels after ischemia/reperfusion might contributes to the cardiac remodeling process, cytosolic to nuclear translocation of the nuclear factor of activated T cells (NFAT) was examined. NFAT is a transcription factor involved in cardiac hypertrophy. Elevated cytosolic [Ca2+]i activates the calmodulin activated serine/threonine protein phosphatase calcineurin which dephosphorylates NFATc resulting in nuclear translocation of NFAT and activation of hypertrophy genes. After ischemia/reperfusion, increased nuclear NFAT was observed (Fig. 4C). When the animals were treated with caspase-8 inhibitor (Q-LETD-OPh) or Rycal (SI 07), NFAT was retained in the cytosol at levels similar to those observed in sham operated animals. This may contribute to the reduction in hypertrophy shown in Fig. 3B. Additionally, mRNA levels of the heart failure marker ANF were also significantly reduced with both treatments (Fig. 4D). In addition to lower levels of collagen (Fig. 3A), interstitial fibrosis as evidenced by increased levels of fibronectin mRNA was also prevented by caspase-8 inhibition or SI 07 inhibition of SR Ca2+ leak (Fig. 4D).
[0583] Ischemia/reperfusion also increased mRNA levels of molecules involved in TNF-a signaling including, TNFR1, TNFR2, caspase-8 and TNF-a (Fig. 4E). This increase in mRNA levels was prevented by the caspase-8 inhibitor (Q-LETD-OPh) or Rycal (SI 07) suggesting a reduced inflammatory response. Taken together, these results indicate that an increase in ROS and NO production, via an early caspase-8 activation, induces RyR2
[0584]S-nitrosylation and diastolic SR Ca2+ leak contributes to I/R injury and long term left ventricular remodeling.
[0586] The therapeutic strategy of rapid reperfusion of ischemic myocardium is designed to preserve cardiac function. However, reperfusion itself has notable adverse effects including arrhythmias and cell death. It is now shown that inhibiting RyR2 mediated diastolic SR Ca2+ leak with a novel orally available drug called rycal (SI 07) which stabilizes the channel, or by
caspase-8 inhibition, significantly reduces reperfusion injury, infarct extension and left ventricular remodeling in the later phase of reperfusion (i.e., 15 days post-reperfusion). LV remodeling following ischemia is caused by multiple factors including: (i) myocardial cell death; (ii) ROS production and inflammatory cytokines; (iii) structural changes of
[0587]myocardium in response to mechanical stress; and (iv) myocardial fibrosis. The present study reports the novel finding that inhibition of RyR2 mediated diastolic SR Ca2+ leak prior to the reperfusion is sufficient to substantially reduce reperfusion injury, myocardial cell death, fibrosis, left ventricular remodeling, and inflammation.
[0588] The results of this study suggests a novel TNF-a mediated signaling pathway wherein caspase-8 activation leads to S-nitrosylation of RyR2 and calstabin2 depletion from the channel complex. The subsequent increase in diastolic SR Ca2+ leak contributes to
[0589]reperfusion injury and left ventricular remodeling after acute ischemia/reperfusion.
[0590] TNFRl is a death-receptor which activates initiator caspases including caspase-8. The resulting activation of caspase-8 is either sufficient to trigger the proteolytic activation of other caspases (i.e., caspase-3), or requires the proteolytic activation of pro-apoptotic proteins of the Bcl2 family, in particular, Bid, which triggers a loss of mitochondrial inner membrane potential Δψιη and ROS generation. In cardiomyocytes, caspase-8 inhibition prevented TNF-a-induced loss of Δψιη and mitochondrial release of cytochorme c. Alternatively, the TNF/TNFR1 complex is thought to regulate sphingolipid signaling pathways. After TNF-a binds to TNFRl an early weak recruitment of FADD and stimulation of caspase-8 in the cell are sufficient to activate sphingomyelinase. Activation of sphingomyelinase initiates sphingolipid metabolism with ceramide, sphingosine and sphingosine-1 -phosphate formation and permits death-receptor oligomerization and caspase-8 activation. These bioactive phospholipids induce cellular responses, such as mitochondrial ROS production and NO synthesis. Hence, early inhibition of caspase-8 prevents TNF-a induced mitochondrial dysfunction and NO production (Fig. 1). Concomitant ROS production and NO production would affect cellular signaling most likely through peroxynitrite formation and
[0591]S-nitrosylation. Oxidation of thiols on RyR2 may activate the channels and under
[0592]pathological conditions this may result in a diastolic SR Ca2+ leak. It is now shown that S-nitrosylation and diastolic SR Ca2+ leak are associated with calstabin2 depletion from the channel complex which is prevented by pharmacological inhibition of caspase-8 or with Rycal SI 07 treatment. Of note only caspase-8 inhibition prevented both calstabin2 depletion and S-nitrosylation. This suggests that changes in the redox environment of the channel may lead to calstabin2 depletion and increased RyR2 channel activity under pathological conditions.
Previous studies have shown that reducing agents or antioxidants are able to normalize RyR2 activity and Ca2+ homeostasis in heart failure. SR Ca2+ leak is thought to trigger cellular damage after acute ischemia and reperfusion. Several studies have reported RyR2 dysfunction after ischemia/reperfusion. Ca2+ overload has been reported to play a pathological role after reperfusion and ventricular arrhythmias. Moreover, reperfusion is associated with the recovery of ATP phosphorylation potential, which restores SR Ca2+-ATPase activity and increases Ca2+ sequestration into the SR. SR Ca2+ overload can cause oscillations of cytosolic Ca2+. Short-term oscillations in cytosolic Ca2+ have been implicated in the genesis of reperfusion arrhythmias. Indeed, caspase-8 inhibition which prevents early RyR2 dysfunction has a profound impact on reperfusion arrhythmias (Fig. 2). In addition, these results suggest a possible connection between circulating TNF-a levels and arrhythmias in acute ischemia as previously suggested. During ischemia reperfusion, aberrant intracellular Ca2+ leak is taken up by the mitochondria. Ca2+ accumulation in the mitochondria leads to activation of mitochondrial permeability transition pore (MPTP). MPTP activation dissipates \|/m and leads to ATP depletion and loss of mitochondrial function. Immediately after MPTP activation mitochondria swell and release apoptogenic factors like cytochrome c and AIF, which activates caspase-dependant and independant execution of apoptosis.
[0593] The present study points out a potential involvement of a dual detection mechanisms sensitizing large scale MPTP opening and mitochondrial membrane permeabilization.
[0594]Accordingly, this sensitizing mechanism suggests that caspase-8-induced mitochondrial depolarization alone, without SR Ca2+ leak, will not be sufficient to trigger cell death but would require a commitment [Ca2+]mito oscillations. Discrete modification of the SR Ca2+ leak may thus be sufficient to prevent large scale swelling and allowing functional recovery of the mitochondria. The present study suggests that leaky RyR2 may contribute to mitochondrial Ca2+ accumulation during ischemia reperfusion and to an amplification loop leading to reperfusion injury given that inhibiting calstabin2 dissociation from the RyR2 complex reduces RyR2 mediated SR Ca2+ leak and is protective. Improving SR Ca2+ handling thus appears to be an potential novel target for reducing reperfusion injury, independently of reducing arrhythmia occurrence per se. In conclusion, the present study highlights the pathophysiological roles of TNF-a-induced caspase-8 activation and ROS/NO production, in the control of RyR2 function after acute myocardial infarction. Early reperfusion induced S-nitrosylation of RyR2 and calstabin2 depletion from the channel complex. Caspase-8 activation also participates in reperfusion injury. Thus, both caspase-8 inhibition and
[0595]RyR2/calstabin2 normalization are potential targets for the prevention of the effects of
reperfusion, including myocardial cell death, arrhythmias and late left ventricular remodeling after acute myocardial infarction.
[0596]It should be understood that various changes and modifications to the methods and compositions described herein are possible without departing from the spirit and scope of the invention. Variations and modifications that can be made without departing from the spirit and scope of the invention will be apparent to those skilled in the art, and all such variations and modifications are within the scope of the invention. For example, further variations and modifications of the invention may be made in accordance with the description provided in U.S. patent applications 09/568,474, 10/288,606, 10/608,723, 10/680,988, 10/763,498, 10/794,218, 10/809,089, 11/088,058, 11/088,123, 11/212,309, 11/506,285, and 11/212,413, and International application PCT/US2006/32405, the contents of which are hereby incorporated by reference in their entirety.
[0598] 1. Defer, N., Azroyan, A., Pecker, F. & Pavoine, C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J Biol Chem 282, 35564-35573 (2007).
[0599] 2. Krown, K.A., et al. TNF alpha receptor expression in rat cardiac myocytes: TNF alpha inhibition of L-type Ca2+ current and Ca2+ transients. FEBS Lett 376, 24-30 (1995).
[0600] 3. Torre- Amione, G et al. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation 92, 1487-1493 (1995).
[0601] 4. Sack, M.N., Smith, R.M. & Opie, L.H. Tumor necrosis factor in myocardial hypertrophy and ischaemia~an anti-apoptotic perspective. Cardiovasc Res 45, 688- 695 (2000).
[0602] 5. Coletta, A., Thackray, S., Nikitin, N. & Cleland, J.G. Clinical trials update: highlights of the scientific sessions of The American College of Cardiology 2002: LIFE, DANAMI 2, MAD IT -2, MIRACLE-ICD, OVERTURE, OCTAVE, ENABLE 1 & 2, CHRISTMAS, AFFIRM, RACE, WIZARD, AZACS, REMATCH, BNP trial and HARDBALL. Eur J Heart Fail 4, 381-388 (2002).
[0603] 6. Coletta, A.P., Clark, A.L., Banarjee, P. & Cleland, J.G. Clinical trials update:
[0604]RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail 4, 559-561 (2002).
[0605] 7. Coletta, A.P., Louis, A.A., Clark, A.L., Nikitin, N. & Cleland, J.G. Clinical trials update from the European Society of Cardiology: CARMEN, EARTH, OPTIMAAL, ACE, TENHMS, MAGIC, SOLVD-X and PATH-CHF II. Eur J Heart Fail 4, 661-666 (2002).
[0606]8. Mann, D.L., et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 109, 1594-1602 (2004).
[0607] 9. Ashkenazi, A. & Dixit, V.M. Death receptors: signaling and modulation. Science 281, 1305-1308 (1998).
[0608] 10. Kroemer, G., Galluzzi, L. & Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 87, 99-163 (2007).
[0609] 11. Luo, X., Budihardjo, I., Zou, H., Slaughter, C. & Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481-490 (1998).
[0610] 12. Zhu, J., Liu, M., Kennedy, R.H. & Liu, S.J. TNF-alpha-induced impairment of mitochondrial integrity and apoptosis mediated by caspase-8 in adult ventricular myocytes. Cytokine 34, 96-105 (2006).
13. Barsacchi, R., et al. Cyclic GMP-dependent inhibition of acid sphingomyelinase by nitric oxide: an early step in protection against apoptosis. Cell Death Differ 9, 1248-1255 (2002).
[0611] 14. Bulotta, S., Barsacchi, R., Rotiroti, D., Borgese, N. & Clementi, E. Activation of the endothelial nitric-oxide synthase by tumor necrosis factor-alpha. A novel feedback mechanism regulating cell death. J Biol Chem 276, 6529-6536 (2001).
[0612] 15. Zima, A.V. & Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71, 310-321 (2006).
[0613] 16. Xu, L., Eu, J.P., Meissner, G. & Stamler, J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234-237 (1998).
[0614] 17. Yan, Y., et al. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res 77, 432-441 (2008).
[0615] 18. Yano, M et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation 112, 3633-3643 (2005).
[0616] 19. Bellinger, A.M., et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15, 325-330 (2009).
[0617] 20. Fauconnier J, et al. (2010) Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 107, 1559-1564.
[0618] 21. Dzietko, M., et al. A critical role for Fas/CD-95 dependent signaling pathways in the pathogenesis of hyperoxia-induced brain injury. Ann Neurol 64, 664-673 (2008).
[0619] 22. Maciejewski, J.P., et al. Nitric oxide suppression of human hematopoiesis in vitro. Contribution to inhibitory action of interferon-gamma and tumor necrosis factor-alpha. J Clin Invest 96, 1085-1092 (1995).
[0620] 23. Kojima, H., et al. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal Chem 70, 2446-2453 (1998).
[0621] 24. Pacher, P., Beckman, J.S. & Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87, 315-424 (2007).
[0622] 25. Lakatta, E.G. & Guarnieri, T. Spontaneous myocardial calcium oscillations: are they linked to ventricular fibrillation? J Cardiovasc Electrophysiol 4, 473-489 (1993).
[0623] 26. Lakireddy, V., et al. The kinetics of spontaneous calcium oscillations and
[0624]arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm 3, 58-66 (2006).
27. Zucchi, R., Ronca, F. & Ronca-Testoni, S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther 89, 47-65 (2001).
[0625] 28. Zucchi, R., et al. Effect of ischemia and reperfusion on cardiac ryanodine receptors— sarcoplasmic reticulum Ca2+ channels. Circ Res 74, 271-280 (1994).
[0626] 29. Passier, R., et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest 105, 1395-1406 (2000).
[0627] 30. Molkentin, J.D. Calcineurin and beyond: cardiac hypertrophic signaling. Circ Res 87, 731-738 (2000).
[0628] 31. Wilkins, B. J. & Molkentin, J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322, 1178-1191 (2004).
[0629] 32. Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol 14, 170- 175 (2005).
[0630] 33. Yellon, D.M. & Hausenloy, D.J. Myocardial reperfusion injury. N Engl J Med 357, 1121-1135 (2007).
[0631] 34. Hori, M. & Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res 81, 457-464 (2009).
[0632] 35. Peng, J., Gurantz, D., Tran, V., Cowling, R.T. & Greenberg, B.H. Tumor necrosis factor-alpha-induced ATI receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res 91, 1119-1126 (2002).
[0633] 36. Peter, M.E. & Krammer, P.H. The CD95 (APO-l/Fas) DISC and beyond. Cell Death Differ 10, 26-35 (2003).
[0634] 37. Scaffidi, C, et al. Two CD95 (APO-l/Fas) signaling pathways. Embo J 17, 1675-1687 (1998).
[0635] 38. Schulze-Osthoff, K et al. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 267, 5317-5323 (1992).
[0636] 39. Hannun, Y.A. Functions of ceramide in coordinating cellular responses to stress. Science 274, 1855-1859 (1996).
[0637] 40. Rotolo, J.A., et al. Caspase-dependent and -independent activation of acid
[0638]sphingomyelinase signaling. J Biol Chem 280, 26425-26434 (2005).
[0639] 41. Lafont, E et al. Caspase-mediated inhibition of sphingomyelin synthesis is involved in FasL-triggered cell death. Cell Death Differ 17, 642-654.
42. Suematsu, N., et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 107, 1418- 1423 (2003).
[0640] 43. Finkel, M.S., et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257, 387-389 (1992).
[0641] 44. Belevych, A.E., et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 84, 387-395 (2009).
[0642]45. Brooks, W.W., Conrad, C.H. & Morgan, J.P. Reperfusion induced arrhythmias following ischaemia in intact rat heart: role of intracellular calcium. Cardiovasc Res 29, 536- 542 (1995).
[0643] 46. Kihara, Y. & Morgan, J.P. Abnormal Cai2+ handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am J Physiol 261, H1746-1755 (1991).
[0644] 47. London, B et al. Calcium-dependent arrhythmias in transgenic mice with heart failure. Am J Physiol Heart Circ Physiol 284, H431-441 (2003).
[0645] 48. Xiao, H., et al. Positive correlation of tumor necrosis factor-alpha early expression in myocardium and ventricular arrhythmias in rats with acute myocardial infarction. Arch Med Res 39, 285-291 (2008).
[0646] 49. Ruiz-Meana, M., Abellan, A., Miro-Casas, E., Agullo, E. & Garcia-Dorado, D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol 297, H1281-1289 (2009).
[0647] 50. Halestrap, A.P. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34, 232-237 (2006).
[0648] 51. Murphy, E. & Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88, 581-609 (2008).
[0649] 52. Piper, H.M., Meuter, K. & Schafer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann Thorac Surg 75, S644-648 (2003).
[0650] 53. Berger, A.B., Sexton, K.B. & Bogyo, M. Commonly used caspase inhibitors designed based on substrate specificity profiles lack selectivity. Cell Res 16, 961-963 (2006).
[0651] 54. Pereira, N.A. & Song, Z. Some commonly used caspase substrates and inhibitors lack the specificity required to monitor individual caspase activity. Biochem Biophys Res Commun 377, 873-877 (2008).
[0652]- I l l -
55. Chauvier, D., Ankri, S., Charriaut-Marlangue, C, Casimir, R. & Jacotot, E.
[0653]Broadspectrum caspase inhibitors: from myth to reality? Cell Death Differ 14, 387-391 (2007).
[0654] 56. Fauconnier, J., et al. Insulin potentiates TRPC3 -mediated cation currents in normal but not in insulin-resistant mouse cardiomyocytes. Cardiovasc Res 73, 376-385 (2007).
[0655] 57. Fauconnier, J., et al. Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes 56, 1136- 1142 (2007).
[0656] 58. Wehrens XH, Lehnart SE, Reiken SR, & Marks AR (2004) Ca2+/calmodulin- dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 94, e61-70.

 (Formula I)
(Formula I)  -CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6;
-CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6; 




 , pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof, or any combination thereof.
, pharmaceutically acceptable salts, hydrates, solvates, complexes and pro-drugs thereof, or any combination thereof. 
 (Formula I)
(Formula I)  -CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6;
-CH2X, acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl; wherein each acyl, alkyl, alkenyl, aryl, alkylaryl, cycloalkyl, cycloalkylalkyl, heteroaryl, heterocyclyl, and heterocyclylalkyl may be optionally substituted, and wherein z is 1, 2, 3, 4, 5, or 6;