[1]Изобретение относится к способам получения высокочистых порошков наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита (далее Сад-КГА), которые могут быть использованы для производства медицинских материалов, стимулирующих восстановление дефектов костной ткани, в том числе в стоматологии, и в качестве сорбентов для адсорбции ионов тяжелых металлов.
[2]В настоящее время синтетический гидроксиапатит Ca10(PO4)6OH2 (далее ГА) и материалы на его основе находят широкое применение в стоматологии, реконструктивно-восстановительной костной хирургии, ортопедии в качестве заменяющего материала для поврежденных зубов и костей [Suchanek W., Yoshimura M. Processing and properties of HA-based biomaterials for use as hard tissue replacement implants //J. Mater. Res. Soc. 1998. V.13. №1. P.94-103]. Многочисленные исследования подтверждают биологическую совместимость ГА с организмом, а также его близкие показатели физических и химических свойств [Bauer I W, Li S. P., Han Y. C. and Yin L. M. Internalization of hydroxyapatite nanoparticles in liver cancer cells //J Mater Sci: Mater Med. 2008. V.19. P.1091-1095.; Abdel-Gawad E. I. and Awwad S. Biocompatibility of Intravenous Nano Hydroxyapatite in Male Rats //Nat. and Sci.2010.V.8. P.60-68].
[3]Благодаря высокой адсорбционной способности по отношению к ионам тяжелых металлов в настоящее время материалы на основе ГА представляют большой интерес к использованию в качестве сорбентов для решения таких экологических проблем как очистка сточных вод, утилизация промышленных отходов, восстановление загрязненных почв [Janga, S. H., Jeonga,Y.G., Mina, B.G., Lyoob, W.S. and Leea, S.L. Preparation and lead ion removal property of hydroxyapatite polyacrylamide composite hydrogels //J. Hazardous Materials. 2008. V.159. P.294-3002]. Кроме того, гидроксиапатиты могут успешно применяться для извлечения ионов тяжелых металлов из крови и восстановления функциональности печени и почки [Removal of Lead Nitrate Toxicity //Journal of American Science. 2011. Vol.7. №.1. P. 105-119 Abdel-Gawad E.I., Awwad S.A. In-vivo and in-vitro prediction of the efficiency of Nano-Synthesized Material in].
[4]Было установлено, что ГА в живых организмах не находится в чистом виде, а всегда содержит в своей структуре примеси, преимущественно карбонат-ионы, благодаря их способности адаптироваться к постоянно меняющимся условиям внутренней среды. Карбонатные группы, содержащиеся в кости (около 4-8% по массе), находятся в нестабильном положении, замещая либо ОН--группы (А-тип замещения), либо (РО4)3--группы (В-тип замещения). Для костной ткани характерен смешанный АВ-тип замещения [Баринов С.М., Комлев В.С. Биокерамика на основе фосфатов кальция. М.: Наука 2005. 204c]. По результатам исследований ряда работ выяснено, что в костях и зубах ГА не является чистым, стехиометрическим Ca10(PO4)6(OH)2 с молярным соотношением Ca/P=10/6=1,67 [Elliott, J. C. Structure and chemistry of the apatites and other calcium orthophosphates //Elsevier: Amsterdam, Holland. 1994. 404p; Dorozhkin, S. V. Calcium orthophosphates //J. Mater. Sci. 2007. V.42. P.1061-1095; Dorozhkin, S. V. Calcium orthophosphates in nature, biology and medicine //Materials. 2009. V.2. P.399-498].
[5]В основном можно считать, что апатит биологического происхождения является кальций-дефицитным карбонатсодержащим с молярным соотношением Ca/(P+CO3)<1,67.
[6]В связи с вышеизложенным синтез высокобиосовместимого наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита для медицинских целей, а также в качестве высокоэффективного сорбента ионов тяжелых металлов для детоксикации организма человека и восстановления окружающей среды является весьма актуальным.
[7]Известен способ получения карбонатсодержащего гидроксиапатита (далее КГА) по патенту EP 0722772 A1, опубликованному 24.07.1996 г., путем нагрева стехиометрического ГА в среде углекислого газа из атмосферного воздуха при примерно 900°С в течение несколько дней. Это трудно контролируемый процесс, который, с одной стороны, приводит к получению КГА с низким коэффициентом замещения, с другой стороны, не всегда получают однофазные образцы. При этом карбонат-анионы замещаются в неоптимальные позиции т.е. в А-позиции, а не в В-позиции, что соответствует получению биомиметического материала подобного биоапатиту.
[8]Известен также способ получения КГА методом осаждения из водных растворов, описанный в источнике J. C. Merry, I. R. Gibson, S. M. Best, W. Bonfield. Synthesis and characterization of carbonate hydroxyapatite (Journal of materials science: materials in medicine. 1998. V.9. P.779-783.), где в качестве источника ионов кальция и фосфат-ионов использовали четырехводный нитрат кальция - Са(NO3)2·4H2O и гидрофосфат аммония - (NH4)2HPO4. Реакцию проводили при температуре 20°С, а рН поддерживали на уровне ≥ 11 за счет добавления раствора гидроксида аммония. Источником карбонат-ионов являлся гидрокарбонат натрия NaHCO3, который добавлялся к раствору гидрофосфата аммония до синтеза. Синтез осуществляли добавлением по каплям смеси растворов гидрофосфата аммония и гидрокарбоната натрия к раствору нитрата кальция. Полученную суспензию ставили для старения на 24 часа. Затем осадок тщательно промывали дистиллированной водой, отфильтровали под вакуумом с последующим высушиванием и измельчением. Недостатками данного способа являются низкая термостабильность продукта при обжиге/спекании, т.е. потеря больших количеств ионов карбоната при нагревании, и трудность за контролем степени замещения карбоната. Кроме того, полученные частицы КГА имеют большой размер и, следовательно, низкую удельную поверхность, а также происходит негативное дополнительное замещение ионов Na+или NH4+в структуре гидроксиапатита.
[9]В патенте EP 0722773 A1 (опубликован 24.07.1996 г.) описан метод получения КГА А-типа, у которого карбонат-анионы замещались только по позиции ОН-групп в структуре ГА. К недостатку данного метода относится несходство по структуре полученных образцов к биоапатиту природной кости.
[10]В патенте EP 0625490 A1 (опубликован 23.11.1994 г.) предложен метод получения КГА, который имеет молярное соотношение Са/Р близко к 1,66. В качестве источника ионов СО32-использовали Na2CO3. Недостаток данного метода состоит в том, что вместе с карбонат-ионами в кристаллической решетке ГА идет нежелательное примесное замещение катионов Na+.
[11]В патенте WO1994008458 A1 (опубликован 28.04.1994 г.) описан способ получения КГА, где исходные реагенты смешивали так, чтобы сформировать гидроксиапатитный цемент при комнатной или физиологической температуре. Источником карбонат-ионов является твердый карбонат кальция СаСО3. Полученный материал представляет собой низкокристаллический или аморфный апатит, который содержит нежелательные ионы натрия.
[12]Патент EP 0342932 A1 (опубликован 23.11.1989 г.) описывает способ получения КГА путем добавления Ca(OH)2 и СаСО3 к суспензии CaHPO4. Ионы CO32-, которые вводятся в реакционную смесь, находятся в виде нерастворимого CaCO3, а не в виде раствора. Молярное соотношение Са/Р согласно этому патенту всегда меньше 1,67. После спекания при температуре от 1000°С до 1100°С содержание карбоната полученного материала уменьшается менее 0,1%. Недостаток данного метода заключается в том, что полученный продукт содержит в своем составе примесь CaCO3 и высокая температура спекания.
[13]В патенте WO 2001083367 A2 (опубликован 08.11.2001 г.) получали КГА путем выдерживания кальция фосфата в сверхкритическом или конденсированном диоксиде углерода. Недостатком данного способа являются техническая сложность и крайне высокая стоимость, которые не позволяют организовать массовое производство продукта.
[14]Наиболее близким по технической сущности является способ, описанный в патенте US 6582672B1 (опубликован 24.06.2003 г. ). Указанный способ получения монофазного КГА включает синтез КГА методом осаждения при рН 10,5-11 путем смешения водных растворов реагентов, содержащих ионы СО32-, РО43- и Са2+. Причем реагенты вводятся в реакционную смесь при условии обеспечения молярного соотношения Са/Р выше 1,67, содержание СО32-, замещенного в Б-позиции или в А и Б позициях, не должно быть выше 5%. Углекислый газ барботируют в деионизированную воду в течение 30 минут, затем к этому раствору прибавляют ортофосфорную кислоту. В полученную смесь при постоянном перемешивании добавляют по каплям суспензию Са(ОН)2 со скоростью 5,5 мл/мин, рН реакционной смеси поддерживают в интервале 10,5-11 путем добавления концентрированного аммиака. Полученную суспензию после синтеза продолжают перемешивать в течение 2 часов и затем оставляют для старения около 12 часов. Вся реакция проходит при комнатной температуре. Осадок фильтруют, промывают дистиллированной водой для удаления избытка аммиака с последующим высушиванием при 80°С в течение 12 часов. После чего высушенный фильтр-корж извлекают, измельчают в мелкий порошок. Частицы порошка имеют средний размер ниже 100 мкм. Это способ позволяет получать монофазовый карбонатсодержащий гидроксиапатит, не содержащий ионы Na+, NH4+. Основными недостатками данного способа являются низкое содержание карбонат-ионов, трудность точного контроля степени замещения СО32-, а кристаллохимическая формула полученного карбонатсодержащего гидроксиапатита не соответствует формуле кальций-дефицитного гидроксиапатита. Кроме того, частицы КГА, полученные данным методом, имеют большой средний размер около 100 мкм.
[15]Задачей данного изобретения является разработка способа получения монофазного наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита наиболее близкого по химическому составу и структуре к природной кости с высокими биоактивностью и удельной поверхностью при молярном соотношении Са/(Р+СО32-)<1,67.
[16]Технический результат:
[17]- получение монофазного продукта Сад-КГА наиболее близкого по химическому составу и структуре к природной кости формулы Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения карбоната в интервале от 0,76 до 1,21, а массовое содержание карбонат-ионов от 5% до 8%;
[18]- в полученном Сад-КГА молярное соотношение Са/(Р+СО32-)<1,67, средний размер кристаллов от 8 нм до 70 нм, удельная поверхность 90-200 м2/г, что обеспечивает повышенную биоактивность и адсорбционную способность готового продукта.
[19]Для решения поставленной задачи получения монофазного наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита предложен способ получения Сад-КГА, включающий синтез путем осаждения из водного раствора реагентов, содержащих ионы СО32-, РО43- и Са2+, отстаивание при комнатной температуре для завершения процесса фазообразования, выделение осадка и высушивание до постоянной массы с последующем измельчением.
[20]Способ включает следующие новые признаки:
[21]- готовят водный раствор композиции гидроксид кальция/карбонат аммония с использованием 0,08-0,16%-ного водного раствора гидроксида кальция и расчетного количества карбоната аммония для получения готового продукта с массовым содержанием карбонат-ионов от 5 до 8 мас.% и молярным соотношением Са/(Р+СО32-) ниже 1,67;
[22]- приливают раствор ортофосфорной кислоты со скоростью 0,5-5 мл/мин на литр водного раствора к композиции гидроксид кальция/карбонат аммония;
[23]- рН реакционной смеси поддерживают выше 11.
[24]В качестве поставщика ионов СО32-использовали карбонат аммония, который позволяет более точно контролировать содержание СО32-в полученном продукте и вносит вклад в поддержание значения рН реакционной смеси.
[25]Количество карбонат-ионов в продукте составляет от 5 до 8 мас.%, что соответствует оптимальному содержанию карбонат-ионов в природной кости. При этом не требуется проведения контроля содержания карбонат-ионов, что обусловливается точной навеской (NH4)2CO3.
[26]Скорость приливания 10-20%-ного раствора ортофосфорной кислоты менее 0,5 мл/мин на литр водного раствора композиции гидроксида кальция и карбоната аммония нецелесообразна из-за крайне большой длительности процесса синтеза.
[27]Скорость приливания 10-20%-ного раствора ортофосфорной кислоты более 5 мл/мин на литр водного раствора композиции гидроксида кальция и карбоната аммония приводит к возможности получения продукта с большой дефектностью в структуре, т.к. при быстром приливании кислоты карбонат-ионы не успевают встраиваться в структуру гидроксиапатита, большая часть их адсорбируется на поверхности образовавшихся кристаллов.
[28]Молярное соотношение Са/(Р+СО32-) выше или равно 1,67, нецелесообразно из-за того, что не дает возможность получить кальций-дефицитный карбонатсодержащий гидроксиапатит и может приводить к появлению побочных продуктов, например СаСО3, Са3(РО4)2.
[29]Если молярное соотношение Са/(Р+СО32-) меньше 1,67 вызывается не из-за дефицита Са, а из-за увеличения содержания замещенных карбонат-ионов в дополнительных позициях, то структура полученного гидроксиапатита не соответствует структуре природной кости.
[30]Значение рН реакционной смеси, поддерживающееся выше 11 благодаря использованию насыщенного раствора гидроксида кальция и карбоната аммония, обеспечивает большое количество гидроксильных групп, что позволяет получить продукт с более завершенной структурой.
[31]Теоретическое обоснование предложенного изобретения заключается в следующем.
[32]Карбонатсодержащие кальций-дефицитные гидроксиапатиты могут быть представлены формулой:
[33]Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения СО32-.
[34]При этом наличие 5-8 мас.% карбоната в составе костной ткани является принципиально важным для формирования полноценной естественной кости с требуемыми строением и функционалом. Для обеспечения необходимого содержания карбоната и кальция в синтетическом карбонатсодержащем гидроксиапатите степень замещения СО32- в синтезируемом продукте должна быть в пределах х=0,76-1,21, а молярное соотношение Са/(Р+CO32-) ниже 1,67.
[35]В предлагаемом способе источником карбонат-ионов является (NH4)2CO3, который добавляют в раствор гидроксида кальция. В результате диссоциации в реакционную смесь поступают свободные карбонат-ионы по реакции:
[36](NH4)2CO3→2NH4++CO32-(1)
[37]Далее свободные ионы CO32-сразу вступают в реакцию с избытком ионов Са2+с образованием малорастворимого карбонат кальция по реакции:
[38]Са2++СО32- → СaCO3↓ (2)
[39]и тем самым уходят из реакционного объема.
[40]Затем в реакционную смесь начинают с заданной скоростью дозировать раствор ортофосфорной кислоты. В этот период происходит формирование промежуточного продукта - аморфного фосфата кальция
[41]3Са2++2PО43- → Сa3(PO4)2↓ (3)
[42]Аморфный фосфат кальция затем трансформируется в термодинамически наиболее устойчивое и самое малорастворимое соединение в системе:
[43]гидроксиапатит СаО-Р2O5-Н2O.
[44]Поскольку CaCO3 является наиболее растворимым соединением в этом ряду, то по мере формирования новой фазы Са3(РO4)2 и далее ГА карбонат кальция начинает растворяться с высвобождением в объем раствора ионов CO32-. Новообразующаяся фаза фосфата кальция, адсорбируя карбонат-ионы и гидроксильные ионы, образует карбонатсодержащий гидроксиапатит.
[45]СaCO3↓ → Са2++СО32- (4)
[46](3-x/2)Сa3(PO4)2+(1-d+3x/2)Са2++xСО32-+(2+x-2d)OH- → Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d(5)
[47]Предложенный способ характеризуют следующие фигуры.
[48]Фиг. 1. Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+СО32-)=1,50, полученного при степени замещения карбоната х=0,76 и содержании СО32-равном 5 мас.%;
[49]Фиг. 2. Ик-спектр образца Сад-КГА с молярным соотношением Са/(Р+СО32-)=1,50, полученного при степени замещения карбоната х=0,76 и содержании СО32-равном 5 мас.%;
[50]Фиг. 3. Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+СО32-)=1,67, полученного при степени замещения карбоната х=1,21 и содержании СО32-равном 8 мас.%.
[51]Фиг. 1 и 3 были получены на дифрактометре Rigaku Ultima IV (Япония) с детектором D/teX Ultra. Съемку проводили в кварцевых кюветах в режиме на отражение (геометрия Брегга-Брентано) с использованием Cu Kα-излучения (длина волны λ=1.54178  ). Параметры работы генератора: ускоряющее напряжение 40 кВ, ток трубки 250 мА. Параметры съемки интервал углов 2θ=5-85°, шаг по 2θ=0.02°, скорость регистрации спектров 3°/мин. Качественный анализ полученных рентгенограмм и профильный анализ спектров, определение значений параметров решетки проводили с помощью программы PDXL Qualitative Analysis при использовании баз данных ICDD (PDF 2008).
). Параметры работы генератора: ускоряющее напряжение 40 кВ, ток трубки 250 мА. Параметры съемки интервал углов 2θ=5-85°, шаг по 2θ=0.02°, скорость регистрации спектров 3°/мин. Качественный анализ полученных рентгенограмм и профильный анализ спектров, определение значений параметров решетки проводили с помощью программы PDXL Qualitative Analysis при использовании баз данных ICDD (PDF 2008).
[52]Фиг. 2 была снята на ИК-Фурье спектрометре Nicolet 6700 (Thermo Electron Corporation, США) с детектором МСТ-А (50 мкм). ИК-спектры поглощения образцов регистрировали в диапазоне 400-4000 см1 со следующими параметрами: число сканов пробы 32; число сканов 32; разрешение 4,000; усиление 8,0; скорость зеркала 0,6329; диафрагма 100,00. Анализ полученных ИК-спектров, определение значений волновых чисел проводили с помощью программного комплекса OMNIC (версия 7.3) при использовании автофильтра, базовой коррекции.
[53]Описание способа поясняется примерами.
[55]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)5,24(CO3)0,76(ОН)0,76 с молярным соотношением Са/(Р+CO32-)-1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната x=0,76, что соответствует около 5 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
[56]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1753 г (NH4)2CO3.
[57]Затем 11,69 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 0,5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[58]Дифрактограмма РФА полученного образца Сад-КГА с молярным соотношением Са/(Р+CO32-)=1,50, полученного при степени замещения карбоната х=0,76, что соответствует 5 мас.% CO32-, представлена на фиг. 1, ИК-спектр образца Сад-КГА с молярным соотношением Са/(Р+CO32-)=1,50 и х=0,76 представлен на фиг. 2.
[60]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)5(CO3)1(ОН)1 с молярным соотношением Са/(Р+CO32-) - 1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната х=1,0, что соответствует около 6,5 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 20 мас.%.
[61]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2306 г (NH4)2CO3.
[62]Затем 5,54 мл 20%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[64]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9(PO4)4,79(CO3)1,21(ОН)1,21 с молярным отношением Са/(Р+CO32-) - 1,50, степенью дефицитности Са2+ d=1 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,08 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
[65]Навеску гидроксида кальция массой 0,8 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,08%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1394 г (NH4)2CO3.
[66]Затем 5,35 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/(NH4)2CO3, со скоростью 3,5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[68]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)5,24(CO3)0,76(ОН)1,48 с молярным соотношением Са/(Р+CO32-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната х=0,76, что соответствует около 5,0 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 15 мас.%.
[69]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,1685 г (NH4)2CO3.
[70]Затем 7,31 мл 15%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[72]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)5(CO3)1(ОН)1,72 с молярным соотношением Са/(Р+CO32-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната x=1,0, что соответствует около 6,5 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
[73]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2218 г (NH4)2CO3.
[74]Затем 10,75 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 5 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм
[76]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы Са9,36(PO4)4,79(CO3)1,21(ОН)1,93 с молярным соотношением Са/(Р+CO32-) - 1,56, степенью дефицитности Са2+ d=0,64 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
[77]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2683 г (NH4)2CO3.
[78]Затем 10,29 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 2 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[80]Синтез наноразмерного кальций-дефицитного карбонатсодержащего гидроксиапатита формулы с молярным соотношением Са/(Р+CO32-) - 1,67 и степенью замещения карбоната х=1,21, что соответствует около 8,0 мас.% CO32-, в случае, когда концентрация водного раствора гидроксида кальция - 0,16 мас.%, а концентрация водного раствора ортофосфорной кислоты - 10 мас.%.
[81]Навеску гидроксида кальция массой 1,6 г при комнатной температуре добавляли к 1000 мл дистиллированной воды, перемешивали с помощью магнитной мешалки в течение 10-15 минут и оставляли на 6 часов до полного растворения гидроксида кальция. После чего к 1000 мл 0,16%-ного водного раствора гидроксида кальция при непрерывном перемешивании добавляли 0,2511 г (NH4)2CO3.
[82]Затем 9,64 мл 10%-ного раствора H3PO4 помещали в делительную воронку и добавляли по каплям в водный раствор, содержащий композицию гидроксид кальция/карбонат аммония, со скоростью 1 мл/мин. После добавления всего объема раствора ортофосфорной кислоты проверяли рН смеси реагентов и рН оставался более 11, в связи с использованием насыщенного раствора Са(ОН)2 и (NH4)2CO3 в качестве исходных реагентов. Реакционную смесь перемешивали в течение 30 минут, а затем оставляли для созревания около 24 часов. Вся реакция проходила при комнатной температуре. Образовавшийся коллоидный раствор отфильтровывали с помощью фильтровальной бумаги. Затем осадок с фильтра количественно переносили в фарфоровую чашку и сушили при 105°С в сушильном шкафу до постоянной массы. После этого Сад-КГА измельчали в мелкий порошок с использованием ступки и пестика и просеивали через сито с диаметром ячейки 0,14 мм.
[83]Дифрактограмма РФА образца Сад-КГА с молярным соотношением Са/(Р+CO32-)=1,67, полученного при степени замещения карбоната х=1,21, что соответствует 8 мас.% CO32-, представлена на фиг. 3.
[84]Физико-химические характеристики образцов, полученных по примерам 1-7, приведены в табл.1.
[85]Таблица 1
Физико-химические характеристики образцов, полученных по примерам 1-7 |
| Образец | Массовая доля СО32-, % | Степень замещения СО32-, х | Молярное соотношение Са/(Р+СО32-) | Количество фаз | Параметры решетки, нм |
| Ось - а | Ось - с |
| Пример 1 | 5 | 0,76 | 1,50 | 1 | 9,418 | 6,901 |
| Пример 2 | 6,5 | 1,0 | 1,50 | 1 | 9,424 | 6,905 |
| Пример 3 | 8 | 1,21 | 1,50 | 1 | 9,415 | 6,908 |
| Пример 4 | 5 | 0,76 | 1,56 | 1 | 9,428 | 6,906 |
| Пример 5 | 6,5 | 1,0 | 1,56 | 1 | 9,434 | 6,895 |
| Пример 6 | 8 | 1,21 | 1,56 | 1 | 9420 | 6,911 |
| Пример 7 | 8 | 1,21 | 1,67 | 2 | 9,397 | 6,916 |
[86]Текстурные характеристики образцов, полученных по примерам 1-7,
[88]Таблица 2
Текстурные характеристики образцов, полученных по примерам 1-7 |
| Образец | Средний размер кристаллов, нм | Удельная поверхность, м2/г | Кристалличность, % |
| Пример 1 | 42,3 | 151,2 | 92,3 |
| Пример 2 | 35,7 | 201,3 | 94,7 |
| Пример 3 | 7,95 | 167,4 | 90,5 |
| Пример 4 | 36,8 | 165,8 | 91,2 |
| Пример 5 | 73,9 | 174,0 | 87,8 |
| Пример 6 | 24,6 | 184,2 | 89,8 |
| Пример 7 | 17,7 | 91,0 | 92,0 |
[89]Определение удельной поверхности, объема и среднего размера пор материалов по методу БЭТ проводили на автоматизированной сорбционной установке TriStar II 3020 производства Micromeritics (США) с использованием объемного варианта сорбционного метода. Удельная поверхность была рассчитана по изотерме низкотемпературной сорбции паров азота по одноточечному методу БЭТ в точке P/Po=0,3189. Образцы были выдержаны в инертном газе азота и гелия с одновременным обеспечением нагрева образцов при температуре 95°С.
[90]Известно, что поверхностный заряд наноразмерных частиц, наряду с их размером и формой, являются важными характеристиками ГА, определяющими процессы клеточной адсорбции. В этом аспекте частицы гидроксиапатита, обладающие отрицательно заряженной поверхностью, имеют заметное преимущество [Yingchao Han et al. Nanosize and Surface Charge Effects of Hydroxyapatite Nanoparticles on Red Blood Cell Suspensions //ACS Appl. Mater. Interfaces. 2012. V.4. №9. P. 4616-4622]. Измерение величины и знака дзета-потенциала частиц КГА после созревания осуществляли методом электрофореза на анализаторе Zetasizer Nano ZS фирмы Malvern Instruments.
[91]Значения дзета-потенциала частиц КГА в виде гидрогеля после 24 часов выдерживания в маточном растворе (время созревания) представлены в табл.3
[92]Таблица 3.
Дзета-потенциал образцов КГА |
| Образец | Молярное соотношение Са/(Р+СО32-) | рН после синтеза | рН после созревания | Значение дзета-потенциала, мВ. |
| Пример 1 | 1,50 | 11,60 | 11,10 | -27,8 |
| Пример 5 | 1,56 | 11,62 | 11,13 | -15,7 |
| Пример 7 | 1,67 | 11,76 | 11,33 | +12,3 |
[93]Проблема загрязнения окружающей среды ионами тяжелых металлов в настоящее время особенно значима. Соединения тяжелых металлов являются распространенными компонентами выбросов транспорта и многих предприятий различных отраслей промышленности. Среди ионов тяжелых металлов наиболее опасными загрязнителями считаются ионы свинца, так как его техногенное накопление в окружающей среде идет высокими темпами. Путем миграции по пищевым цепям ионы Pb2+попадают в организм человека, вызывая единовременные или хронические отравления, и приводят к серьезным нарушениям процессов обмена веществ и жизненно важных функций организма [Herbert L. Needleman, Christine McFarland, Roberta B. Ness, Stephen E. Fienberg, Michael J. Tobin. Bone lead levels in adjudicated delinquents A case control study //J. Neurotoxicology and Teratology. 2002. V.24. P. 711-717; Sharma A., Sharma V., Kansal L. Amelioration of lead-induced hepatotoxicity by Allium sativum extracts in Swiss albino mice //Libyan J. of Med. 2010. V.5. P. 4621-4630]. Одним из наиболее опасных последствий действия ионов свинца считается его способность замещать кальций в костях и быть постоянным источником отравления в течение длительного времени [Silbergeld E.K. Facilitative mechanisms of lead as a carcinogen //Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis. 2003. V.533. P. 121-133].
[94]Сорбцию ионов свинца на полученных образцах изучали при комнатной температуре при 25±2°С с использованием ацетатного буферного раствора при рН=5,5. В качестве поставщика ионов Pb2+был выбран Pb(CH3COO)2·3H2O. В химические стаканы емкостью 100 мл, содержащие навески 0,1±0,0010 г исследуемых образцов, помещали по 50 мл модельных растворов с разными концентрациями ионов Pb2+в диапазоне концентраций Pb2+от 40 до 4800 мг/л. Определение равновесных концентраций ионов свинца и ионов кальция в модельных растворах было проведено после 48 часов экспозиции.
[95]Концентрацию ионов свинца определяли на вольтамперометрическом анализаторе АКВ-07МК с твердотельным электродом (компания Аквилон, Россия). Для определения концентрации ионов свинца была использована методика с пределом обнаружения 0,2 мг/дм3; диапазон тока - 5000 мА; время накопления - 10 с.
[96]Данные о сорбционной емкости полученных образцов, представленные в табл.4, определили графическим путем, строя график зависимости отношения равновесной концентрации ионов свинца С к величине сорбции Г от величины сорбции Г, используя традиционное
[97]уравнение Лэнгмюра Г=V(Co−C)m и его преобразованное уравнение в линейной формеCГ=1kГо+СГо
и его преобразованное уравнение в линейной формеCГ=1kГо+СГо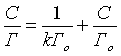
[98]где Г- величина сорбции, мг/г; V - объем раствора ионов свинца, мл; C - равновесная концентрация ионов свинца, мг/л; Cо - исходная концентрация ионов свинца, мг/л; Го - сорбционная емкость, мг/г; k - константа адсорбции, характеризующая степень сродства данного иона к сорбции по Ленгмюру.
[99]Таблица 4
Сорбционная емкость образцов, полученных по примерам 1-6, по отношению к ионам тяжелых металлов (при рН=5,5) |
| Образец | Массовая доля СО32-, % | Молярное соотношение Са/(Р+СО32-) | Сорбционная емкость к ионам Pb2+, мг/г |
| Пример 1 | 5 | 1,50 | 1689 |
| Пример 2 | 6,5 | 1,50 | 1724 |
| Пример 3 | 8 | 1,50 | 1720 |
| Пример 4 | 5 | 1,56 | 1697 |
| Пример 5 | 6,5 | 1,56 | 1686 |
| Пример 6 | 8 | 1,56 | 1650 |
[100]Из табл.4 следует, что в зависимости от степени дефицитности и содержания карбоната сорбционная емкость синтетических образцов Сад-КГА по отношению к ионам Pb2+составляет до 1720 мг/г, что превышает почти в 5 раз сорбционную емкость немодифицированного гидроксиапатита [Bailliez S., Nzihou А., Beche А., Flamant G. Removal of lead (Pb) by hydroxyapatite sorbent //Trans IChemE, Part B, Process Safety and Environmental Protection. 2004.V. 82. №2.Р. 175-180].
[101]Таким образом, приведенные примеры подтверждают возможность осуществления предложенного способа с достижением заявленного технического результата:
[102]- получение монофазного продукта Сад-КГА наиболее близкого по химическому составу и структуре к природной кости формулы Ca10-d(PO4)6-x(CO3)x(OH)2+x-2d, где d - степень дефицитности Са2+; х - коэффициент или степень замещения карбоната в интервале от 0,76 до 1,21, а массовое содержание карбонат-ионов от 5% до 8%;
[103]- повышенную биоактивность и адсорбционную способность в полученном Сад-КГА с удельной поверхность 90-200 м2/г, что обеспечивается за счет молярного соотношения Са/(Р+СО32-)<1,67 и среднего размера кристаллов от 8 нм до 70 нм.

