[1]Изобретение относится к новым биологически активным производным 1-(1-адамантил)этиламина (римантадина) и может найти применение в фармакологии и вирусологии. Предлагаемые синтезированные адамалтил-производные в виду их значительной активности и низкой токсичности можно предложить в качестве кандидатов новых противовирусных препаратов
[2]Проблема гриппа - одна из актуальнейших научных задач последнего столетия. Вирусы гриппа активно циркулируют в природе. Актуальность гриппа как медицинской проблемы обусловлена высокой заболеваемостью и смертностью пациентов. На долю гриппа и гриппоподобных заболеваний приходится до 90% всех инфекций. Пандемия 2009/2011 года, вызванная вирусом гриппа A(H1N1)pdm2009 года, показала как крайнюю ограниченность, так и недостаточную эффективность существующих лекарственных средств. ВОЗ сообщает, что по состоянию на 2 октября 2014 года в мире насчитывалось 668 случая (подтвержденных лабораторными исследованиями) инфицирования человека высоковирулентным вирусом гриппа птиц A/H5N1, 394 из которых закончились летальным исходом, при этом уже многие годы летальность от этой инфекции сохраняется высокой. Сохраняется и опасность появления в природе смертельных для человека мутантов вируса гриппа A/H5N1. Последние данные, свидетельствующие о возможности получения таких вариантов в лабораторных условиях, вызвали серьезную тревогу в мире [1].
[3]Поэтому создание новых препаратов, действующих на широкий диапазон штаммов вируса гриппа, в том числе и высоко вирулентных, является неотложной и одной из первостепенных задач здравоохранения.
[4]Первыми эффективными противогриппозными препаратами были на основе аминопроизводных адамантанового карбоцикла: римантадина гидрохлорида - ремантадин и 1-аминоадамантана - амантадин. Ингибирующее действие этих соединений направлено на угнетение функции протон-проводящего канала М2 в оболочке вируса. В настоящее время методами кристаллографии установлено, что четыре субъединицы этого белка образуют в мембране вируса канал, по которому осуществляется транспорт протонов через мембрану вирусной частицы. Попавшие во внутрь вириона протоны запускают процесс диссоциации вирусных белков и выход генетического материала вируса в цитоплазму клетки. Механизм противовирусного действия ремантадина и амантадина состоит в том, чтобы нарушать нормальный ток протонов. В результате химического прессинга ремантадина на вирусы гриппа А к нему возникла резистентность, достигающая 90%. Потерю активности в основном связывают с мутацией в трансмембранном домене белка М2 вируса гриппа.
[5]В настоящее время для лечения и профилактики гриппа используют ингибиторы нейраминидазы - Озельтамивир (Тамифлю) и Занамивир (Реленза), они действуют на этапе выхода вновь синтезированных вирионов вируса гриппа из оболочки клетки, путем блокирования отщепления вирусных частиц от поверхности клеток. Однако известны случаи формирования резистентности к озельтамивиру, в частности практически вся популяция сезонных штаммов вируса гриппа A/H1N1 была резистентна к озельтамивиру [2, 3].
[6]Ранее нами был предложен путь присоединения большого числа функциональных групп к молекуле римантадина гидрохлорида [4]. Этот путь открывает новые возможности для создания эффективных препаратов прямого действия. При появлении новых генотипов вируса гриппа А со структурно измененными белками вируса позволяет быстро, а главное синтетически и экономически доступно вносить изменения в имеющееся адамантил-производное соединение для преодоления резистентности. Важно, что при распаде этих соединений в организме будут образовываться хорошо изученный в биологическом аспекте римантадин гидрохлорид и остатки природных аминокислот.
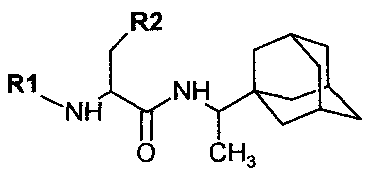
[7]Сущность изобретения заключается в создании новых синтетических соединений, являющихся производными 1-(1-адамантил)этиламина, а именно трет-бутилоксикарбонил-L-метионинсульфонил-1-адамантаилэтиламин (далее соединение 1), L-метионинсульфонил-1-адамантаилэтиламин хлоргидрат (далее соединение 2) и карбобензилоксикарбонил-L-триптофанил-1-адамантаилэтиламина (далее соединение 3), которые обладают противовирусной активностью в отношении высоко патогенных штаммов вируса гриппа А и ингибируют репродукцию штаммов, резистентных к действию римантадина гидрохлорида. Предлагаемые соединения 1 и 3 обладают вирулицидным действием по отношению к этим штаммам вируса гриппа. Соединения 1, 2 и 3 имеют следующие структурные формулы:
[8]
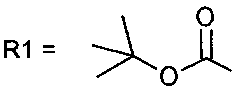
[9]Соединение 1,  ;
; 
[10]Соединение 2, R1= -Н; 
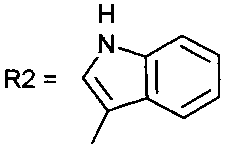
[11]Соединение 3,  , а
, а 
[12]Более того, соединения обладают меньшим токсическим эффектом на монослой клеток Madin Darby Canine Kidney (MDCK) и клеток почек эмбриона свиньи (СПЭВ), чем римантадин гидрохлорид.
[13]Для получения соединений 1, 2 и 3 может быть использованы различные подходы, например: карбодиимидный метод, азидный метод, методом активированных эфиров и т.д. Получить соединения 1, 2 и 3 можно также методом смешанных ангидридов (фиг. 1). Однако предложенный метод синтеза не должен рассматриваться как некое ограничение объема настоящего изобретения во всех отношениях.
[14]Технический результат - получены новые, малотоксичные, соединения, обладающие избирательной противогриппозной активностью в отношении штаммов вируса гриппа А, в том числе резистентных к действию римантадина гидрохлорида. Предлагаемые синтезированные адамалтил-производные в виду их значительной активности и низкой токсичности можно предложить в качестве кандидатов новых противовирусных препаратов.
[15]Краткое описание чертежей. Для более ясного понимания сути заявленного изобретения, которое отражено в формуле изобретения, а также для демонстрации ее особенностей и преимуществ далее приводится подробное описание со ссылками на фигуры чертежей.
[16]На фиг. 1 представлена схема синтеза производных 1-(1-адамантил)этиламина методом смешанных ангидридов на примере соединения I.
[17]На фиг. 2 представлены в виде таблицы 1 физико-химические константы соединений 1, 2 и 3.
[18]На фиг. 3 представлены в виде таблицы 2 данные влияния различных концентраций соединений 1, 2 и 3 на репродукцию пандемического штамма вируса гриппа A/IIV-Orenburg/83/2012(H1N1)pdm09 в культуре клеток MDCK при добавлении вещества одномоментно с вирусом. Испытания противовирусной активности соединений 1, 2 и 3 проведены в сравнении с римантадином гидрохлоридом.
[19]На фиг. 4 представлены в виде таблицы 3 данные противовирусных свойств соединений 1, 2 и 3 в отношении инфекции, вызванной высоко патогенным вирусом гриппа A/duck/Novosibirsk/56/05 (H5N1), а так же цитотоксическое действие этих соединений на монослой клеток СПЭВ.
[20]На фиг. 5 представлены в виде таблицы 4 данные вирулицидной активности синтезированных соединений в отношении высокопатогенного вируса гриппа A/duck/Novosibirsk/56/05 (H5N1).
[21]Один из способов получения соединений 1, 2 и 3 - метод смешанных ангидридов (фиг. 1). Образование пептидной связи между римантадином гидрохлоридом, содержащим аминогруппу, и аминокислотами, защищенными по аминогруппе трет-бутилоксикарбонильной (Вос-) или бензилоксикарбонильной (Z-) группой, проводили в одну стадию в эквимолярном соотношении.
[22]При синтезе соединений использовали рацемический римантадин гидрохлорид фирмы Zhejiang Kangyu Pharmaceutical Со (Китай), L-аминокислоты фирмы Sigma-Aldrich (США). Использовали изо-бутилхлорформиат (TBCF) фирмы «Fluka» (Швейцария), N-метилморфолин (NMM) фирмы Sigma-Aldrich (США). Все используемые для конденсации и удаления защитных групп растворители предварительно абсолютировали и перегоняли по стандартным методикам. Идентификация полученных соединений осуществлялась с помощью тонкослойной хроматографии (ТСХ) на пластинах Silufol (Чехия) в системах: метанол-хлороформ, 13:60 (А), втор-бутанол - 3%-ный аммиак, 100:44 (В), н-бутанол - уксусная кислота - вода - пиридин, 30:3:12:10 (С), позволяющих констатировать полное отсутствие в испытуемых образцах следов римантадина гидрохлорида. Молекулярный вес был установлен на MALDI-TOF-времяпролетном масс-спектрометре Bruker UltraFlex II с программным обеспечением для сбора и обработки масс-спектров flexControl 1.1. и flexAnalys 2.2. Инфракрасные спектры были получены на ИК Фурье спектрометр ИнфраЛЮМ ФТ-10. Удельное оптическое вращение полученных соединения определяли в стандартных условиях на автоматическом поляриметре А1-ЕПЛ (1%-ный раствор в этиловом спирте, длина кюветы 0,5 дм). Температуру плавления полученных соединений измеряли на приборе «SMP 20» Stuart Scientific (Великобритания). Для идентификации аминокислотных остатков в полученных соединениях проводили кислотный гидролиз в 6 н HCl при 105°C в течении 12 часов. Образующиеся свободные аминокислоты идентифицировали с помощью ТСХ в системах фенол - вода, 1:1 и н-бутанол - уксусная кислота - вода, 3:2:2.
[23]Для более конкретного понимания биологических свойств полученных соединений в предлагаемом изобретении под противовирусной и вирулицидной активности нужно понимать следующее.
[24]Вирулицидная активность соединения связана с прямым инактивирующим действием на вирионы в составе вирусной популяции, в результате чего частично или полностью утрачивается инфекционная активность вируса. Чтобы проверить вирулицидные свойства вещества (соединения), достаточно провести инкубацию смеси вируса и вещества в течение определенного времени, после чего проверить инфекционные свойства вируса без исследуемого вещества и вируса в смеси с веществом методом титрования в культурах клеток или в организме лабораторных животных. Достоверное снижение инфекционной активности вируса на 1,0 и более логарифмов (lg) или ее полная утрата по сравнению с вирусом без вещества свидетельствует о проявлении вирулицидной активности исследуемого соединения.
[25]Противовирусная активность соединения, как правило, включает как вирулицидную активность вещества, так и его противовирусные свойства, т.е. способность ингибировать ту или иную стадию репликации вируса, от адсорбции вируса на клетки, его проникновения до влияния на сборку и выход вируса из зараженной клетки. Поэтому противовирусную активность исследуемого вещества проверяют путем обработки монослоя клеток или введением вещества лабораторным животным до заражения вирусом (профилактический эффект вещества), в момент заражения (лечебно-профилактический эффект) и через определенный интервал времени после заражения (лечебный эффект). Противовирусный эффект вещества оценивается как по проценту жизнеспособных инфицированных клеток, или здоровых лабораторных животных, сохранившихся в условиях применения вещества, по сравнению с контрольной группой, так и по его свойству снижать способность инфицированных клеток или зараженных животных продуцировать инфекционный вирус.
[26]Настоящее изобретение проиллюстрировано нижеследующими примерами. Однако эти примеры не должны рассматриваться как некое ограничение объема настоящего изобретения во всех отношениях.
[27]Пример 1. Синтез соединения 1 (Вос-Met(SO2)-Rem)
[28]Синтез осуществляется в две стадии.
[29]1) Синтез Boc-Met(SO2)-OH (трет-бутилоксикарбонил-метионинсулъфон) и его выделение.
[30]1,0 г (5,5 мМ) метионинсульфона и 0,22 г (5,5 мМ) NaOH растворили в 5,0 мл Н2О и 4,0 мл трет-бутилового спирта. При перемешивании при 45°C тремя порциями в течении 1 часа прибавляют 1,4 г (6,42 мМ) ди-трет-бутилдикарбоната (пирокарбонат). По окончанию реакции на роторном испарителе в вакууме 15 мм. р. ст. и температуре 50°C отгоняют трет-бутиловый спирт, остаток разбавляют водой в 1,5 раза, и экстрагируют гексаном (15,0 мл × 3). Затем водный раствор подкисляют раствором 1,36 г KHSO4 в 5 мл H2O до рН 3-3,5.
[31]Полученный раствор в делительной воронке экстрагируют этил ацетатом (15,0 мл × 4). Этилацетатные экстракты сушат безводным Na2SO4, растворитель удаляют в роторном испарителе при вакууме 15 мм. р. ст. при 50°C. В остатке получается масло, которое впоследствии кристаллизуется в белые кристаллы. Выход кристаллического продукта 1,37 г (88%), Тпл. = 112°C, Rf=0,55 (А); Rf=0,81 (B); Rf=0,78 (С), [α]20D=+8∘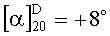 (с=1, спирт).
(с=1, спирт).
[32]2) Синтез Boc-Met(SO2)-Rem (трет-бутилоксикарбонил-1-метионинсулъфонил-адамантаилэтиламина) и его выделение, (см фиг. 1)
[33]В трехгорлую колбу, снабженную механической мешалкой, термометром и хлоркальцевой трубкой вносят 1,0 г (3,5 мМ) трет-бутилоксикарбонил-метионинсульфон (Boc-Met(SO2)-OH) в 10,0 мл. CHCl3 с 15,0 мл тетрагидрофурана и прибавляют 0,4 мл (3,5 мМ) N-метилморфолина (NMM). Охлаждают до -25°C и при перемешивании в реакционную массу добавляют 0,47 мл (3,5 мМ) изо-бутилхлорформиата (IBCF). Перемешивают 10 мин. Затем добавляют заранее приготовленный и охлажденный до -20°C хлоргидрат 1-(1-адамантаил)этиламина 0,77 г (3,5 мМ) в 10 мл CHCl3 с 0,4 мл (3,5 мМ) NMM. Перемешивают - 30 мин при -20-15°C, затем еще 1 час при 0°C и 10 часов при комнатной температуре.
[34]Реакционную массу переносят в колбу и растворитель (CHCl3) удаляют на роторном испарителе при 50°C и 15 мм. р. ст. Остаток растворяют в 35,0 мл этилацетата и 10,0 мл H2O. Раствор переносят в делительную воронку и последовательно промывают, 0,5 н серной кислотой (4,0 мл×1), 0,5 н KHCO3 (10,0 мл × 2), затем подкислили 1н H2SO4 и промывают Н2О (5,0 мл × 1). Органический слой отделяют и сушат безводным Na2SO4. Этилацетат удаляют в вакууме, получают вспененное масло, которое при промывке диэтиловым эфиром твердеет.
[35]Выход: 1,31 г. (83%), Тпл. = 152-154°C Rf=0,90 (A); Rf=0,89 (B); Rf=0,91 (С), [α]20D=+7∘ (с=1, спирт).
(с=1, спирт).
[36]Соединение 2 является аналогом соединения 1, в котором аминогруппа деблокирована от трет-бутилоксикарбонильной защитной группировки по следующей методике:
[37]Синтез соединения 2 (HCl*H-Met(SO2)-Rem, 1-метионинсулъфонил-адамантаилэтиламин хлоргидрат) и его выделение.
[38]К раствору 1,0 г (2,26 мМ) Boc-Met(SO2)-Rem в 10,0 мл этилацетата при 5°C прибавляют 4,4 мл этилацетата насыщенного 4 н HCl. Реакционную смесь выдерживают в течение 1 часа при 20°C, периодически помешивая. Контроль за прохождением реакции ведут по ТСХ. По завершению реакции реакционную смесь высаждают диэтиловым эфиром. Растворители декантируют. Остаток сушат в вакууме. Оставшееся масло при растирании в эфире кристаллизуется.
[39]Выход 0,85 г (96%). Тпл. = 138-140°C; [α]D=+6° (с=1, спирт), Rf=0,53 (A), Rf=0,63 (B).
[40]Пример 2. Синтез соединения 3 (Z-Trp-Rem, Карбобензилоксикарбони-L-лтриптофанил-1-адамантаилэтиламин) и его выделение.
[41]Z-Trp-OH (Карбобензилоксикарбонил-L-триптофан) был получен от фирмы Sigma-Aldrich (США). Чистота ≥99.0%, показатель вращения плоскополяризованного света [α]20D=+2.9±0.3∘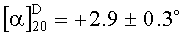 (с=3% в уксусной кислоте), температура плавления 124-127°C. Коммерческий Z-Trp-OH был использован в синтезе конечного соединения.
(с=3% в уксусной кислоте), температура плавления 124-127°C. Коммерческий Z-Trp-OH был использован в синтезе конечного соединения.
[42]В трехгорлую колбу, снабженную механической мешалкой, термометром и хлоркальцевой трубкой вносят 1,0 г (2,95 мМ) карбобензилоксикарбони-L-триптофана (Z-Trp-ОН) в 10,0 мл. CHCl3 с 15,0 мл тетрагидрофурана и прибавляют 0,32 мл (2,95 мМ) N-метилморфолина (NMM). Охлаждают до -25°C и при перемешивании в реакционную массу добавляют 0,40 мл (3,0 мМ) изо-бутилхлорформиата (IBCF). Перемешивают 10 мин. Затем добавляют заранее приготовленный и охлажденный до - 20°C хлоргидрат 1-(1-адамантаил)этиламина 0,63 г (2,95 мМ) в 10 мл CHCl3 с 0,4 мл (3,5 мМ) NMM. Перемешивают - 30 мин при -20 -15°C, затем еще 1 час при 0°C и 10 часов при комнатной температуре.
[43]Реакционную массу переносят в колбу и растворители удаляют на роторном испарителе при 50°C и 15 мм. р. ст. Остаток растворяют в 35,0 мл этилацетата и 10,0 мл Н2О. Раствор переносят в делительную воронку и последовательно промывают, 0,5 н серной кислотой (4,0 мл × 1), 0,5 н KHCO3 (10,0 мл × 2), затем подкислили 1н H2SO4 и промывают Н2О (5,0 мл × 1). Органический слой отделяют и сушат безводным Na2SO4. Этилацетат удаляют в вакууме, получают вспененное масло, которое при промывке диэтиловым эфиром твердеет.
[44]Выход: 1,41 г. (95,5%), аморфный, Rf=0,85 (A); Rf=0,93 (B); Rf=0,69 (С), [α]20D=+5∘ (с=1, спирт).
(с=1, спирт).
[45]Пример 3. Определение противовирусной активности синтетических соединений в отношении вируса гриппа A/IIV-Orenburg/83/2012(H1N1)pdm09
[46]Изучение специфической противовирусной активности синтетических соединений проводили на 96-луночных панелях со сформировавшимся монослоем клеток культуры ткани MDCK. Одномоментно с инфицированием в монослой клеток вносили римантадин гидрохлорид (референс препарат) и изучаемые синтетические соединения в концентрациях 0,05, 0,5, 2,5, 5,0, 7,5 и 10,0 мкг/мл. Панели инкубировали 24 часа при 37°C, а затем останавливали реакцию фиксированием клеток 80% ацетоном на фосфатном буфере. Постановку метода клеточного иммуноферментного анализа (ИФА) проводили согласно методике, описанной ранее [5, 6]. Процент ингибирования вирусной активности соединениями определяли по формуле (1):
[47]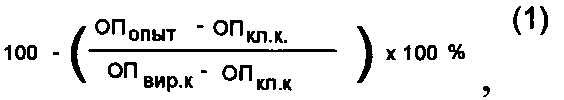
[48]где ОПопыт - оптическая плотность опытной лунки (с веществом) при 492 н.м., ОП кл.к - ОП492 клеточного контроля, ОП вир.к. - ОП492 вирусного контроля.
[49]На фиг. 3 представлены средние значения результатов испытания противовирусной активности синтезированных соединений из параллельных опытов, проводимых в аналогичных условиях. Значения результатов показанных в этих опытах соответствовали нормальному распределению.
[50]Из данных фиг. 3 видно, что синтезированные соединения 1, 2 и 3 защищают клетки монослоя MDCK от цитопатического действия вируса. Соединения 1, 2 и 3 показали значительный процент ингибирования репродукции штамма вируса гриппа A/IIV-Orenburg/83/2012(H1N1)pdm09, устойчивого к действию римантадина гидрохлорида. Ингибирующая доза (ИД50) для соединения 1 составила 5 мкг/мл, а для соединений 2 и 3 7,5 мкг/мл. Отсутствие ингибирующего эффекта римантадина гидрохлорида косвенно свидетельствует о резистентности данного штамма к препаратам адамантанового ряда.
[51]Пример 4. Определение противовирусной активности предлагаемых соединений в отношении высокопатогенного вируса гриппа A/H5N1 и их цитотоксического действия.
[52]Изучение противовирусной активности синтезированных соединений и их цитотоксического действия проводили на панелях со сформировавшимся монослоем СПЭВ. В качестве высокопатогенного вируса гриппа А использовали штамм вируса гриппа A/duck/Novosibirsk/56/05 (H5N1) [7]. Противовирусную активность проверяли в трех схемах введения соединений в культуру клеток: за 6 часа до заражения клеток, в момент заражения и через 6 часа после заражения культур клеток СПЭВ. Соединения вносили в концентрациях 1,0, 0,5, 0,25, 0,12 и 0,06 мг/мл. Оценку цитотоксического действия синтезированных соединений определяли колориметрическим методом после инкубации клеток с предлагаемыми соединениями в них в течение 72 часов при 37°C.
[53]Противовирусный эффект вещества оценивается по проценту жизнеспособных инфицированных клеток, путем сравнения интенсивности окрашивания раствора в контрольных и опытных лунках при добавлении нейтрального красного на автоматическом спектрофотометре при длине волны 450 нм. Концентрация, которая обеспечивала выживание 50% клеток СПЭВ, принималась за величину ИД50 (Фиг. 4).
[54]Токсический эффект предлагаемых соединений оценивается по проценту погибших клеток монослоя. Концентрация, которая вызывала гибель 50% клеток СПЭВ, принималась за величину ЦТ50 (Фиг. 4)
[55]Из данных фиг. 4 видно, соединение 1 было эффективно во всех схемах внесения соединения. Наиболее эффективно соединение 1 при внесении за 6 часов до заражения и одномоментно (одновременно) с вирусом (ИД50<0,06 мг/мл). Соединение 2 было эффективно при внесении соединения 1 за 6 часов до заражения и одномоментно с вирусом (ИД50<0,06 мг/мл), однако при внесении через 6 часов после заражения ингибирующая концентрация уменьшилась вдвое (ИД50<0,12 мг/мл). Соединение 3 также проявляло высокий эффект защиты клеток при всех схемах внесения (ИД50<0,06 мг/мл), а по показателю ЦТ50 это соединение превосходит соединение 1 и 2, т.е. соединение 3 менее токсично, чем соединения 1 и 2.
[56]Пример 5. Исследование вирулицидной активности соединений в отношении вируса гриппа A/duck/Novosibirsk/56/05 (H5N1)
[57]В опыте использованы концентрации соединений 10,0 мг/мл, которые смешивали с вирусом следующим образом - 200,0 мкл раствора соединения с добавлением 100,0 мкл вируссодержащего материала в исходной концентрации. Экспозиция с вируссодержащим материалом проведена при комнатной температуре в течение 20 минут, после чего титровали инфекционную активность вируса в каждом варианте опыта в культурах клеток СПЭВ при различных разведениях смеси соединений с вирусом. По разнице титров вируса о контрольных и опытных экспертиментах судили о вирулицидной активности соединения - его способности подавлять инфекционную активность вируса гриппа A/duck/Novosibirsk/56/05 (H5N1). Вирулицидная активность синтетических соединений проиллюстрирована в таблице 4 (фиг. 5). В опытных экспериментах 200,0 мкл раствора соединения (концентрация 10 мг/мл) смешивали со 100,0 мкл вируссодержащего материала в исходной концентрации. В качестве контроля - вируссодержащий материал без предлагаемого соединения (вещества). Экспозиция 20 мин ТЦИД50/200 мкл - тканевая инфекционная доза вируса, заражающая 50% экспонированных клеточных культур, при внесении 200 мкл раствора предлагаемого соединения.
[58]В результате была обнаружена высокая вирулицидная активность для синтетических соединений 1 и 3. Снижение инфекционного титра составило около четырех логарифмов (1000 раз) по отношению к вирусному контролю. Соединение 2 снижало активность вируса на 1,5 логарифма.
[59]Т.о., получены новые соединения формулы 1, 2 и 3 на основе адамантил-аминокислот, обладающие избирательной противовирусной активностью в отношении высокопатогенных вирусов гриппа А. Соединения 1-3 in vitro обладают меньшей токсичностью по сравнению с ремантадином. Причем соединение 3 имеет наибольшую ЦТ50, по сравнению с соединениями 1 и 2 а также с римантадином гидрохлоридом. Соединения 1 и 3 проявляют вирулицидную активность в отношении высоковирулентного вируса гриппа птиц A/H5N1. Механизм противовирусного действия полученных соединений до конца не ясен. Основной предполагаемый механизм противовирусного действия, вероятно, сходен с действием римантадина гидрохлорида на протонный канал М2 вируса гриппа А [8, 9].
[60]Предлагаемые соединения могут быть применены для создания новых противовирусных препаратов, ингибиторов функции протонных каналов вируса. Причем созданные лекарственные средства на основе предлагаемых соединений могут использоваться как в виде индивидуального лекарства, так и в составе комплексной терапии.
[62]1. Herfst S., Schrauven E.J.A., Linster M., et al. Airborne Transmission of Influenza A/H5N1 Virus Between Ferrets. // Science. - 2012. V. 336. №6088. P. 1534-1541.
[63]2. Львов Д.К., Бурцева Е.И., Галегов Г.А. и др. Чувствительность эпидемических и пандемических штаммов вирусов гриппа к занамивиру (Релензе™) в опытах in vitro // Вопр. вирусол., 2010. 55(6), с. 10-14.
[64]3. Смирнов B.C., Гаршинина А.В., Штро А.А. Протективная активность комбинации глутамил-триптофана и глицирризиновой кислоты при пероральном введении на модели экспериментальной летальной гриппозной инфекции у белых мышей, вызванной осельтамивирустойчивым штаммом вируса. // Вопр. вирусол., 2014. 59(5), с. 31-38.
[65]4. Шибнев В.А., Гараев Т.М., Финогенова М.П. и др. Производные 1-(1-адамантил)этиламина и их противовирусная активность, Патент РФ RU 2461544 С1 приоритет от 19 апреля 2011 г.
[66]5. Бурцева Е.И., Шевченко Е.С., Ленева И.А. и др. Чувствительность к ремантадину и арбидолу вирусов гриппа, вызвавших эпидемические подъемы заболеваемости в России в сезоне 2004-2005 гг. // Вопр. вирусол. 2007. №2. С. 24-29.
[67]6. Ленева И.А., Фадеева Н.И., Федякина И.Т. и др. Применение иммуноферментной индикации вирусспецифических антигенов в изучении нового противовирусного препарата. // Хим. - фарм. журнал. 1994. №9. С. 4-15.
[68]7. Львов Д.К., Щелканов М.Ю., Дерябин П.Г. и др. Метод первичной изоляции штаммов вируса гриппа А, штамм virus A/duck/Novosibirsk/56/05 H5N1 для приготовления диагностических, профилактических и лечебных препаратов, для оценки противовирусной активности различных соединений. Патент РФ RU 2309983 С1 приоритет от 25 ноября 2005 г.
[69]8. Hu F., Luo W., Hong М. «Mechanisms of Proton Conduction and Gating in Influenza M2 Proton Channels from Solid-State NMR». Science, v. 330 no. 6003, p. 505-508 (2010).
[70]9. Balannik V., Carnevale V., Fiorin G., «Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel». Biochemistry v. 49, p. 696-708 (2010).
 ,
, ;
;  ,
, ,
, ,
,  .
.
