[0001] ORGANOSILICON COMPOUNDS AND THEIR USE AS THE MODULATORS OF THE TRPV1 RECEPTOR
[0002]The present invention relates to compounds and their use. In particular, it relates to novel silicon-containing compounds having pharmacological activity, to pharmaceutical compositions containing them, and to their use in therapy. The compounds function as modulators at the TrpVl receptor and therefore have potential use in the treatment or prevention of conditions having an association with the vanilloid receptor 1, TrpVl.
[0003]The mammalian transient receptor potential (TRP) channels are a superfamily (including TRPC, TRPV, TRPP, TRPM, TRPA, TRPML and TRPN families) of ion channels that have a diverse range of physiological functions and are present in many tissues and almost all cell types. TrpVl (also known as VRl) is a member of the TRPV family represented by TRPV1-TRPV6 in addition to Osm-9 from C. elegans and Nanchung (Nan) from Drosophila. TrpVl was first identified in 1997 (1) as a non-selective cation channel and the target for capsaicin, the active ingredient in hot peppers. TrpVl is activated or sensitized by moderate heat and a number of different inflammatory mediators such as capsaicin, vanilloid, endocannabinoid, eicosanoids, anandamides and low pH. The receptor is highly expressed in small to medium sensory neurons of the dorsal root and trigeminal ganglia (1), and innervates many visceral organs including bone, bladder, dermis, lungs and gastrointestinal tract. It is also expressed in other neuronal and non-neuronal tissues including the CNS nuclei, kidney, stomach and T-cells. Hence, the established function of TrpVl as a sensory mediator has resulted in the development of agonists and antagonists with potential in the treatment of a diverse range of chronic inflammatory conditions, for example neuropathic pain and more recently arthritis (2).
[0004]TrpVl is associated with the transduction of painful thermal stimuli (1) and disruption of the VRl gene in mice resulted in a lack of response to capsaicin-, acid-, and heat-gated responses previously reported in small diameter dorsal root ganglion neurons and impaired thermal inflammatory hyperalgesia (3,4). This research confirms that VRl plays an essential role in human pain pathways and antagonists have utility in the treatment of a diverse range of pain conditions, for example TrpVl antagonist SB-705498 effectively reduces hyperalgesia and allodynia in animal models (5). Expression of TrpVl on sensory nerves that innervate maxillary molar teeth suggests that activation of the channel contributes to tooth pain (6). VRl antagonist compounds e.g. BCTC, GRC-621 1, ABT-102, JNJ-17203212, JNJ-38748021, NGD-8243 and SPM-955 are currently being investigated for use in the treatment of pain, including migraine, dental pain, neuropathic pain and urinary incontinence-associated pain.
[0005]Over-expression of TrpVl in the gastrointestinal tract has been shown in inflammatory bowel disease, Crohn's Disease and ulcerative colitis; therefore receptor activation is implicated in gastrointestinal inflammation and function (7). TrpVl antagonists JNJ-17203212 and JNJ-38748021 are currently in clinical development for the treatment of inflammatory bowel disease, particularly colitis. TrpVl is implicated directly in the pathogenesis of pancreatitis (8) as many of the mediators that participate in the development of the condition also act on TrpVl expressed on the sensory fibres (9).
The ability of capsaicin to induce a cough and the presence of TrpVl on the sensory respiratory nerves together provide strong evidence that TrpVl is implicated in cough induction. Therefore antagonists may have use in the treatment of asthma, cough and chronic obstructive pulmonary disease (COPD) (10, 11). Clinical evidence supports an increase in TrpVl sensitivity to capsaicin-induced cough in patients suffering from respiratory infection or allergic asthma (12, 13). Antagonist compound JNJ- 17203212 is in development for the treatment of cough and has been shown to reduce the number of capsaicin-induced coughs in animal models.
[0006]TrpVl is functionally expressed in the uroepithelium and implicated in the development of the micturition reflex in both normal and pathological conditions (14).
[0007]Antagonists to TrpVl have been shown to have efficacy in models of bladder disorders, for example GRC- 6211 completely abolished increased bladder reflex activity in models of cystitis, supporting the role of TrpVl in bladder inflammation (15).
[0008]TrpVl is expressed on sensory neurons innervating the pancreas and recent research using TrpVl knock out models suggest that antagonists may have benefit in the treatment of type I (automimmune) diabetes (16).
[0009]Furthermore TrpVl is reported as a promising new target for therapeutic interventions in obesity related disorders, and type II diabetes (17).
[0010]WO200208221 (Neurogen) discloses TrpVl antagonist diary 1 piperazines useful in the treatment of chronic and acute pain, urinary incontinence and itch. WO2003066595 (Euroceltique) discloses TrpVl antagonist piperazine compounds useful in the treatment of pain, urinary incontinence, inflammatory conditions and neurodegenerative diseases. WO2008002247 (Astra Zeneca) discloses pyridine P2Yi2 antagonist compounds useful in the treatment of thrombotic conditions and cardiovascular diseases. WO200401 1441 (Euroceltique) discloses pyridazinylpiperazine mGluRl antagonists useful in the treatment of pain, urinary incontinence, inflammatory bowel disease and irritable bowel syndrome.
[0011]There is a continued clinical need for the development of further classes of TrpVl modulators that demonstrate improved drug-like properties.
[0012]Sila-substitution (C/Si-exchange) of drugs is a relatively recent approach for searching for organo-silicon compounds which have beneficial biological properties. The approach involves the replacement of specific carbon atoms in compounds by silicon. A review of this approach is provided in Tacke and Zilch, Endeavour, New Series, 10, 191-197 (1986).
[0013]In accordance with a first aspect of the present invention, there is provided a compound having the Formula (1):
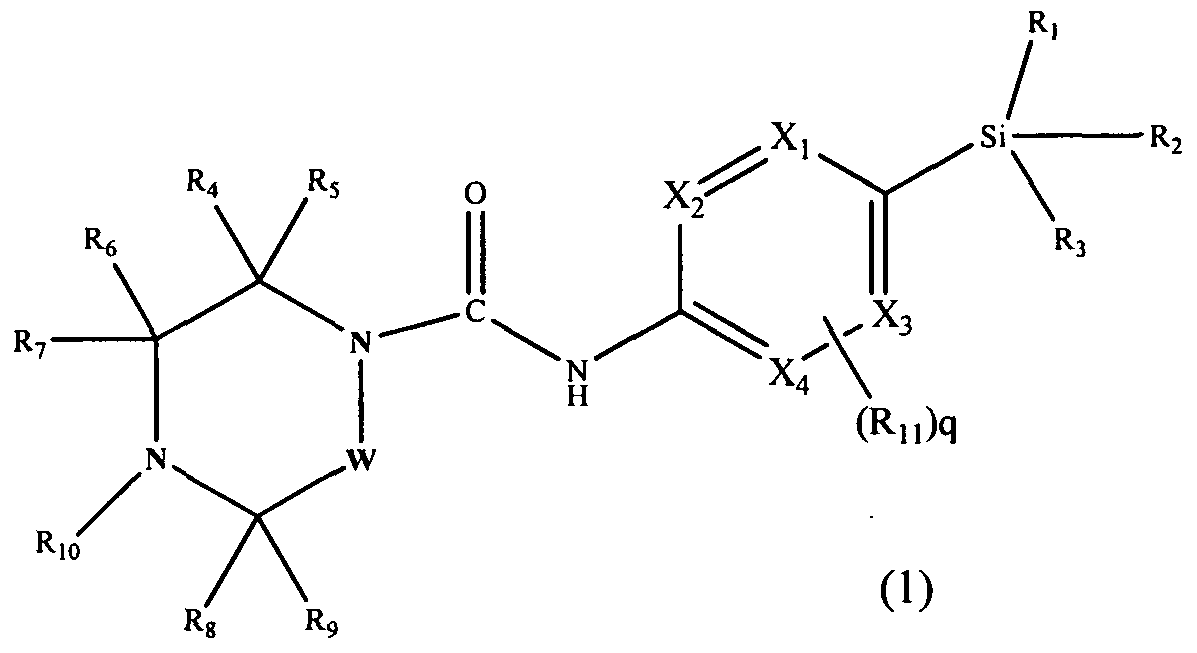
[0015]Xi, X2, X3 and X4 independently represent CH or N;
[0016]R1, R2 and R3 independently represent Ci.6 alkyl, C3-8 cycloalkyl or C3.8 cycloalkylCi_6alkyl, each of which groups may be optionally substituted with one or more substituents independently selected from halogen and haloCi-6 alkyl;
[0017]R4, R5, R6, R7, Re and R9 independently represent H, Ci-6 alkyl, C3-8 cycloalkyl or -Qi-ORi4, or R4 and R9 may join to form a bridging Ci-6 alkylene chain;
[0018]Rio represents aryl or heteroaryl, each of which may be optionally substituted by one or more substituents, independently selected from halogen, Ci-6 alkyl, haloCi_6 alkyl, hydroxyC^alkyl, haloC].6alkoxy, cyano, -Q2-CO2Ri2, -Q2-COR,2, -Q2-CONR12Ri3, -Q2-OR12, -Q2-NR12Rn, -Q2-NR12SO2R,,, -Q2- NRi2CORi3, -Q2-SO2NRi2R13, -Q2-S(O)1nRi2, -Y-aryl, -Y-heteroaryl, -Y-C3-8 cycloalkyl and -Y-heterocyclyl;
[0019]Qi and Q2 independently represent a covalent bond, Ci-6 alkylene, or C1-6 alkylene substituted with hydroxy;
[0020]R12 and R13 independently represent a H atom, or a C1-6 alkyl or C3-8 cycloalkyl group, in which each group may be optionally substituted with one or more Q-6 alkoxy; or when Ri2 and R13 are attached to the same nitrogen atom they may join to form a nitrogen containing heterocyclyl ring, which may be optionally substituted with one or more substituents independently selected from Ci-6 alkyl and C1-6 alkoxy;
[0021]RM represents a H atom, or a Q-6 alkyl or C3-8 cycloalkyl group, in which each group may be optionally substituted with one or more C1-6 alkoxy;
[0022]Y represents a covalent bond, C1-6 alkylene, or -O-. m represents O, 1 or 2;
[0023]Rn, which is optionally present and may be attached to any available carbon atom X1 to X4 instead of H, represents halogen, haloC1-6alkyl, C)-6alkoxy, haloCi-6alkoxy, cyano or Ci-6 alkyl which may be optionally substituted with one or more substituents independently selected from halogen, haloC1-6 alkyl and OR16;
q represents 0, 1 or 2;
[0024]W represents -(CH2),,-, which may be optionally substituted with one or more substituents independently selected from Ci-6 alkyl, C3.8 cycloalkyl and -Q3-ORi5; n represents 1 or 2;
[0025]Q3 represents a covalent bond or C]-6 alkylene; Ri5 represents H, C]-6 alkyl, or C3-8 cycloalkyl; R]6 is as defined for R]4; or a pharmaceutically acceptable salt or ester thereof.
[0026]The compounds of the invention have been found to modulate the TrpVl receptor. In particular, the compounds possess antagonist activity at this receptor. Based on the high affinity for the receptor, the compounds may have the potential to display useful selectivity for the TrpVl receptor.
[0027]Where any group in the compound of formula (I) above is referred to as being optionally substituted, this group may be unsubstituted or substituted by one or more substituents. Typically any such group will be unsubstituted, or substituted by one or two substituents.
[0028]In the compounds of the invention as represented by formula (1) and the more detailed description hereinafter certain of the general terms used in relation to groups and substituents thereon are to be understood to include the following atoms or groups unless otherwise specified.
[0029]The term 'Cx-y alkyl' as used herein refers to a linear or branched saturated hydrocarbon group containing from x to y carbon atoms. For example, C]-6 alkyl refers to a linear or branched saturated hydrocarbon group containing from 1 to 6 carbon atoms. Examples of Ci-6 alkyl groups include methyl, ethyl, n-propyl, isopropyl, n- butyl, isobutyl, sec-butyl, tert butyl, n-pentyl, isopentyl, neopentyl and hexyl.
[0030]The term 'Cx-y alkylene' as used herein refers to a divalent hydrocarbon group obtained by removing one hydrogen atom from 'Cx-y alkyl' above. Examples of Q-6 alkylene groups include methylene, ethylene, propylene, methylmethylene and dimethylmethylene.
[0031]The term 'Cx-y alkoxy' as used herein refers to an -O-Cx-y alkyl group wherein Cx-y alkyl is as defined herein. Examples of such groups include methoxy, ethoxy, propoxy, butoxy, pentoxy and hexoxy.
[0032]The term 'Cx-y cycloalkyl' as used herein refers to a saturated monocyclic hydrocarbon ring of x to y carbon atoms. For example, C3-8 cycloalkyl refers to a saturated monocyclic hydrocarbon ring of 3 to 8 carbon atoms. Examples of C3-8 cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
[0033]The term 'C3-8 CycloalkylCi-6 alkyl' refers to a Ci-6 alkyl group as defined herein wherein a hydrogen atom is replaced with a C3-8 cycloalkyl group. Examples include cyclopropylmethyl.
The term 'halogen' as used herein refers to a fluorine, chlorine, bromine or iodine atom, unless otherwise specified.
[0034]The term "cyano" represents a -CN group. The term "hydroxy" represents a -OH group.
[0035]The term 'hydroxyCi-6 alkyl' as used herein refers to a Q-6 alkyl group as defined herein wherein at least one (preferably one to three) hydrogen atom is replaced with hydroxy. Examples of such groups include hydroxymethyl, hydroxyethyl, dihydroxyethyl, hydroxypropyl and hydroxyisopropyl.
[0036]The term 'haloC)-6 alkyl' as used herein refers to a Ci-6 alkyl group as defined herein wherein at least one (preferably one to three) hydrogen atom is replaced with halogen. Examples of such groups include fluoroethyl, trifluoromethyl and trifluoroethyl.
[0037]The term
 group as defined herein wherein at least one (preferably one to three) hydrogen atom is replaced with halogen. Examples of such groups include trifluoromethoxy and difluoromethoxy.
group as defined herein wherein at least one (preferably one to three) hydrogen atom is replaced with halogen. Examples of such groups include trifluoromethoxy and difluoromethoxy.
[0038]The term 'aryl' as used herein refers to a C6-^ monocyclic or bicyclic hydrocarbon group which comprises at least one aromatic ring. Examples of such groups include phenyl, naphthyl and tetrahydronaphthalenyl.
[0039]The term 'heteroaryl' as used herein refers to a 5-6 membered monocyclic aromatic or a fused 8-10 membered bicyclic aromatic group which comprises monocyclic or bicyclic ring containing 1 to 4 heteroatoms selected from oxygen, nitrogen and sulphur. Examples of such monocyclic aromatic groups include thienyl, furyl, furazanyl, pyrrolyl, triazolyl, tetrazolyl, imidazolyl, oxazolyl, thiazolyl, oxadiazolyl, isothiazolyl, isoxazolyl, thiadiazolyl, pyranyl, pyrazolyl, pyrimidyl, pyridazinyl, pyrazinyl, pyridyl, triazinyl, tetrazinyl and the like. Examples of such bicyclic aromatic groups include quinolinyl, isoquinolinyl, quinazolinyl, quinoxalinyl, pteridinyl, cinnolinyl, phthalazinyl, naphthyridinyl, indolyl, isoindolyl, azaindoly], indolizinyl, indazolyl, purinyl, pyrrolopyridinyl, furopyridinyl, benzofuranyl, isobenzofuranyl, benzothienyl, benzoimidazolyl, benzoxazolyl, benzoisoxazolyl, benzothiazolyl, benzoisothiazolyl, benzoxadiazolyl, benzothiadiazolyl and imidazopyridyl.
[0040]When the 'heteroaryl' contains a nitrogen atom as its ring-constituting atom, the nitrogen atom may be oxidized. For instance, pyridyl as the 'heteroaryl' may be its N-oxide.
[0041]The term 'heterocyclyl' refers to a 4-7 membered monocyclic group or a fused 8-12 membered bicyclic group which may be saturated or partially unsaturated, which monocyclic or bicyclic group contains 1 to 4 heteroatoms selected from oxygen, nitrogen, silicon or sulphur. Examples of such monocyclic groups include pyrrolidinyl, azetidinyl, pyrazolidinyl, oxazolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, hydantoinyl, valerolactamyl, oxiranyl, oxetanyl, dioxolanyl, dioxanyl, oxathiolanyl, oxathianyl, dithianyl, dihydrofuranyl, tetrahydrofuranyl, dihydropyranyl, tetrahydropyranyl, tetrahydropyridinyl, tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, diazepanyl and azepanyl. Examples of such bicyclic groups include indolinyl, isoindolinyl, benzopyranyl, quinuclidinyl, 2,3,4,5-tetrahydro-lH-3- benzazepine and tetrahydroisoquinolinyl.
The term 'nitrogen containing heterocycJyl ring' refers to a ring containing at least one nitrogen atom and selected from rings corresponding to the 'heterocyclyl' groups mentioned above. Examples of such rings include pyrrolidine, azetidine, piperidine, piperazine, morpholine and thiomorpholine.
[0042]'Pharmaceutically acceptable salts' of compounds of Formula (1) of the present invention include but are not limited to acid addition salts (for example, phosphates, nitrates, sulphates, borates acetates, maleates, citrates, fumarates, succinates, methanesulfonates, benzoates, salicylates and hydrohalides), salts derived from inorganic bases (such as lithium, potassium and sodium), salts of amino acids "(such as glycine, alanine, valine, leucine, isoleucine, cysteine, methionine, proline), organic bases (such as triethylamine, hydroxide, choline, thiamine, and N-N'-diacetylethylenediamine). Other pharmaceutically acceptable salts include ammonium salts, substituted ammonium salts and aluminium salts. Further pharmaceutically acceptable salts include quaternary ammonium salts of the compounds of formula I.
[0043]The compound of Formula 1 of the present invention may be in either hydrate or non-hydrate form.
[0044]'Pharmaceutically acceptable esters' of compounds of Formula (1) are derivatives in which one or more carboxyl (i.e. -C(O)OH) groups of the said compounds are modified by reaction with an alcoholic moiety G- OH so as to yield -C(O)OG groups, wherein G may be CM8 alkyl (e.g. Ci-6 alkyl), aryl, heteroaryl, C3-8 cycloalkyl or combinations thereof.
[0045]General methods for the preparation of salts and esters are well known to the person skilled in the art. Pharmaceutical acceptability of salts and esters will depend on a variety of factors, including formulation processing characteristics and in vivo behaviour, and the skilled person would readily be able to assess such factors having regard to the present disclosure.
[0046]Where compounds of the invention exist in different enantiomeric and/or diastereoisomeric forms (including geometric isomerism about a double bond), these compounds may be prepared as isomeric mixtures or racemates, although the invention relates to all such enantiomers or isomers, whether present in an optically pure form or as mixtures with other isomers. Individual enantiomers or isomers may be obtained by methods known in the art, such as optical resolution of products or intermediates (for example chiral chromatographic separation (e.g. chiral HPLC)), or an enantiomeric synthesis approach. Similarly, where compounds of the invention may exist as alternative tautomeric forms (e.g. keto/enol, amide/imidic acid), the invention relates to the individual tautomers in isolation, and to mixtures of the tautomers in all proportions.
[0047]In one embodiment two of the four groups selected from X1, X2, X3 and X4 independently represent N.
[0048]In another embodiment one of the groups selected from Xi, X2, X3 and X4 represents N (e.g. X4)
[0049]In another embodiment X], X2, X3 and X4 each represent CH.
[0050]In one embodiment R], R2 and R3 independently represent Q-6 alkyl (e.g. methyl, ethyl, n-propyl) in which each group may be optionally substituted with one or more (preferably one to three) substituents independently selected from halogen and haloCi_6 alkyl.
[0051]More particularly Ri, R2 and R3 each represent unsubstituted Ci-6 alkyl (e.g. methyl, ethyl, n-propyl).
Even more particularly R1, R2 and R3 each represent unsubstituted methyl.
[0052]In one embodiment R4, R5, R6, R7, R8 and R9 independently represent H or C1-6 alkyl (e.g. methyl). In another embodiment R4 and R9 represent C1-6 alkyl (e.g. methyl) and R5, R6, R7 and R8 represent H. In a further embodiment R4, R5, R6, R7, R8 and R9 each represent H. In yet another embodiment, R4 represents C)-6 alkyl (e.g. methyl) and R5, R6, R7, R8 and R9 represent H.
[0053]In another embodiment R10 represents aryl or heteroaryl, each of which may be optionally substituted by one or more (preferably one to three) substituents, independently selected from halogen, C1-6 alkyl, haloC1-6 alkyl, hydroxyC1-6alkyl, cyano, -Q2-CO2R12, -Q2-COR12, -Q2-CONR12R13, -Q2-OR12, -Q2-NR12R13, -Q2- SO2NR12R13, and -Q2-S(O)mR12.
[0054]Typical examples of aryl or heteroaryl represented by R10 include phenyl, imidazolyl, thiazolyl, pyrimidyl, pyridazinyl, pyrazinyl, pyridyl, quinolinyl and isoquinolinyl. In one embodiment R1O represents optionally substituted phenyl. In another embodiment R]0 represents optionally substituted imidazolyl. In a further embodiment R10 represents optionally substituted thiazolyl. In yet another embodiment R1O represents optionally substituted pyrimidyl. In another embodiment R10 represents optionally substituted pyridazinyl. In a further embodiment R1O represents optionally substituted pyrazinyl. In a still further embodiment R10 represents optionally substituted pyridyl. In another embodiment R]0 represents optionally substituted quinolinyl. In a further embodiment R1O represents optionally substituted isoquinolinyl.
[0055]In a further embodiment Rio represents aryl (e.g. phenyl) which may be optionally substituted by one or more (preferably one to three) substituents, which may be independently selected from halogen (e.g. chloro, bromo, fluoro) and haloC1-6alkyI (e.g. trifluoromethyl).
[0056]In a further embodiment R10 represents heteroaryl (e.g. pyridyl, imidazolyl, thiazolyl, pyrimidyl, pyridazinyl, pyrazinyl, pyridyl, quinolinyl or isoquinolinyl) optionally substituted by one or more (preferably one to three) substituents, which may be independently selected from halogen, C1-6 alkyl, haloC]-6 alkyl, hydroxyC1-6alkyl, cyano, -Q2-CO2R12, -Q2-COR12, -Q2-CONRi2R13, -Q2-OR12, -Q2-NR12R13, -Q2-SO2NR12R13 and -Q2- S(O)1nR12
[0057]In certain embodiments R10 represents pyridyl (e.g. pyridin-4-yl or pyridin-2-yl) pyridazinyl (e.g. pyridazyn- 3-yl), thiazolyl (e.g. thiazol-2-yl), quinolinyl (e.g. quinolin-5-yl), isoquinolinyl (e.g. isoquinolin-5-yl), pyrazinyl (e.g. pyrazin-2-yl), pyrimidyl (e.g. pyrimidin-4-yl) or imidazolyl (e.g. imidazol-2-yl), each of which may be optionally substituted by one to three substituents which may be selected from: halogen (e.g. chloro, fluoro), haloC1-6 alkyl (e.g. trifluoromethyl), C1-6 alkyl (e.g. methyl), hydroxyC1-6alkyl (e.g. hydroxymethyl, hydroxyethyl, 1 -hydroxyethyl, 2-hydroxyisopropyl), cyano, -Q2-CO2R12 (e.g. ethoxycarbonyl, methoxycarbonyl, methoxycarbonylmethyl, carboxy, carboxymethyl), -Q2-COR12 (e.g. isobutyryl), -Q2-CONR12R13 (e.g. carbamoyl, pyrrolidin-1-ylcarbonyl, methylcarbamoyl, dimethylcarbamoyl), -Q2-OR]2 (e.g. methoxymethyl, methoxymethoxymethyl), -Q2-NR12Ri3 (e.g. dimethylaminomethyl), -Q2- SO2NRi2R13 (e.g. dimethylaminosulphonyl) and -Q2-S(O)1nR12 (e.g. methylsulphonyl).
Specific examples of Rio include: phenyl, 2,4-dichlorophenyl, 2-chlorophenyl, 2-bromophenyl, 2-trifluoromethylphenyl, 2-fluorophenyl, 2,3- dichlorophenyl, 2,5-dichlorophenyl, pyridin-4-yl, pyridin-2-yl, 3-methylpyridin-2-yl, 3,5-dichloropyridin-4- yl, 3-chloropyridin-2-yl, 3-methylsulphonylpyridin-2-yl, 3-cyanopyridin-2-yl, thiazol-2-yl, quinolin-5-yl, isoquinolin-5-yl, 3-fluoropyridin-2-yl, 3-trifluoromethylpyridin-2-yl, pyrazin-2-yl, 3-cyanopyrazin-2-yl, 3- chloro-5-(ethoxycarbonyl)pyridin-2-yl, 3-chloro-5-carboxypyridin-2-yl, 3-chloro-5-(carbamoyl)pyridin-2-yl, 3-chloro-5-(pyrrolidine-l-ylcarbonyl)pyridin-2-yl, 3-chloro-5-(hydroxymethyl)pyridin-2-yl, 6-chloro-4- methylpyridazin-3-yl, 3-chloro-5-((dimethylamino)methyl)pyridin-2-yl, 3-(methoxycarbonyl)pyridin-2-yl, 3- carboxypyridin-2-yl, 3-(hydroxymethyl)pyridin-2-yl, 3-(methylcarbamoyl)pyridin-2-yl, 5-methylpyrimidin- 4-yl, 5-cyano-3-methylpyridin-2-yl, 6-chloro-5-methylpyridazin-3-yl, 5-fluoropyrimidin-4-yl, 4- methylpyridazin-3-yl, 3-chloro-5-(methoxymethoxymethyl)pyridin-2-yl, 5-methylpyridazin-3-yl, 3-chloro-5- (dimethylcarbamoyl)pyridin-2-yl, 3-methylpyrazin-2-yl, 3-chloro-5-(methoxymethyl)pyridin-2-yl, 3-chloro- 5-(2-hydroxypropan-2-yl)pyridin-2-yl, 3-chloro-5-(l-hydroxyethyl)pyridin-2-yl, 3-chloro-5- ((methoxycarbonyl)methyl)pyridin-2-yl, 3-chloro-5-(carboxymethyl)pyridin-2-yl, 3-chloro-5-(2- hydroxyethyl)pyridin-2-yl, 3-chloro-5-methylpyridin-2-yl, 3-chloro-5-(dimethylaminosulphonyl)pyridin-2-yl, 3-chloro-5-(ethoxycarbonyl)pyridin-2-yl, 3,5-dichloropyridin-2-yl, 3-chloropyridin-2yl-oxide, 3-methyl-5- (methoxycarbonyl)pyrazin-2-yl, 3-methyl-5-(carbamoyl)pyrazin-2-yl, thiazol-2-yl, 5- (methoxycarbonyl)thiazol-2-yl, 4-(methoxycarbonyl)thiazol-2-yl, 5-carboxythiazol-2-yl, 4-carboxythiazol-2- yl, 4-(hydroxymethyl)thiazol-2-yl, 5-(methylcarbamoyl)thiazol-2-yl, 5-(dimethylcarbamoyl)thiazol-2-yl, 5- (pyrrolidine- 1 -ylcarbonyl)thiazol-2-yl, 4-(methylcarbamoyl)thiazol-2-yl, 4-(dimethylcarbamoyl)thiazol-2-yl, 4-(pyrrolidine-l -ylcarbonyl)thiazol-2-yl, 5-(hydroxymethyl)thiazol-2-yl, 3-fluoro-5- (methoxycarbonyl)pyridin-2-yl, 3-methyl-5-(methoxycarbonyl)pyridin-2-yl, 1 -methyl- 1 H-imidazol-2-yl, 3- fluoro-5-carboxypyridin-2-yl, 3-methyl-5-carboxypyridin-2-yl, 3-chloro-5-(methylsulphonyl)pyridin-2-yl, and 3 -chloro-5 -(isobutyry l)pyridin-2-y 1.
[0058]Rio is typically unsubstituted or mono- or di-substituted. In one embodiment R]0 is unsubstituted. In another embodiment R]0 is monosubstituted. In another embodiment Rio is disubstituted.
[0059]In another embodiment Qi and Q2 independently represent a covalent bond or Ci-6 alkylene. (e.g. methylene, ethylene or propylene).
[0060]In a further embodiment Q2 represents a covalent bond or
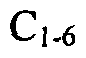 alkylene (e.g. methylene, dimethylmethylene, methylmethylene or ethylene). In a further embodiment Q2 represents a methylene or ethylene linkage.
alkylene (e.g. methylene, dimethylmethylene, methylmethylene or ethylene). In a further embodiment Q2 represents a methylene or ethylene linkage.
[0061]In a further embodiment Q2 represents a covalent bond.
[0062]In one embodiment Ri2 and Rj3 independently represent a H atom or a Ci-6 alkyl group which may be optionally substituted with one or more (preferably one to three) C]-6 alkoxy; or when R]2 and R]3 are attached to the same nitrogen atom they may join to form a nitrogen containing heterocyclyl ring.
[0063]In another embodiment R]2 and Ri3 independently represent H or Ci-6 alkyl (e.g. methyl, ethyl, isopropyl) optionally substituted with one or more (preferably one to three) Ci-6 alkoxy (e.g. methoxy).
In another embodiment R^ and R|3 are attached to the same nitrogen atom and may join to form a nitrogen containing heterocyclyl ring (e.g. pyrrolidine) optionally substituted with Ci_6alkyl (e.g. methyl).
[0064]In a further embodiment Ri2 is H or methyl, methoxymethyl, ethyl or isopropyl.
[0065]In a further embodiment Rn is H or methyl.
[0066]In another embodiment RH represents an H atom or a
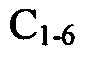 alkyl (e.g. methyl).
alkyl (e.g. methyl).
[0067]In another embodiment Y represents a covalent bond or Ci-6 alkylene (e.g. methylene).
[0068]In another embodiment m represents 2.
[0069]In another embodiment Rn, which is optionally present, may be attached to any available carbon atom, and represents halogen,
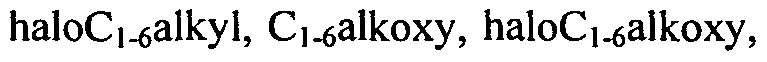 cyano or C1-6 alkyl which may be optionally substituted with one or more (preferably one to three) substituents independently selected from halogen, haloQ.6 alkyl and OR]6.
cyano or C1-6 alkyl which may be optionally substituted with one or more (preferably one to three) substituents independently selected from halogen, haloQ.6 alkyl and OR]6.
[0070]In a further embodiment Rn represents halogen (e.g. chloro, fluoro), haloCi-6alkyl (e.g. trifluoromethyl), cyano or Ci-6 alkyl (e.g. methyl or ethyl).
[0071]In a further embodiment Ri i is attached to X4 or X3 and represents halogen (e.g. chloro or fluoro), ImIoC1.6alkyl (e.g. trifluoromethyl), cyano or Ci- β alkyl (e.g. methyl or ethyl).
[0072]In certain embodiments, q represents 0 or 1. In particular embodiments, q represents 0.
[0073]In one embodiment W represents -(CH2),,- optionally substituted with one or more (preferably one to three) C,-6alkyl.
[0074]In a further embodiment W is unsubstituted -CH2-.
[0075]In one embodiment n represents 1. In another embodiment n represents 2.
[0076]In one embodiment Q3 represents a covalent bond or C^alkylene (e.g. methylene or ethylene).
[0077]In one embodiment R!5 represents H, C]-6 alkyl (e.g. methyl or ethyl).
[0078]In one embodiment Ri6 represents H, Ci.6 alkyl (e.g. methyl or ethyl).
[0079]One subclass of compounds according to the invention is represented by the compounds of formula l(A):-
[0080]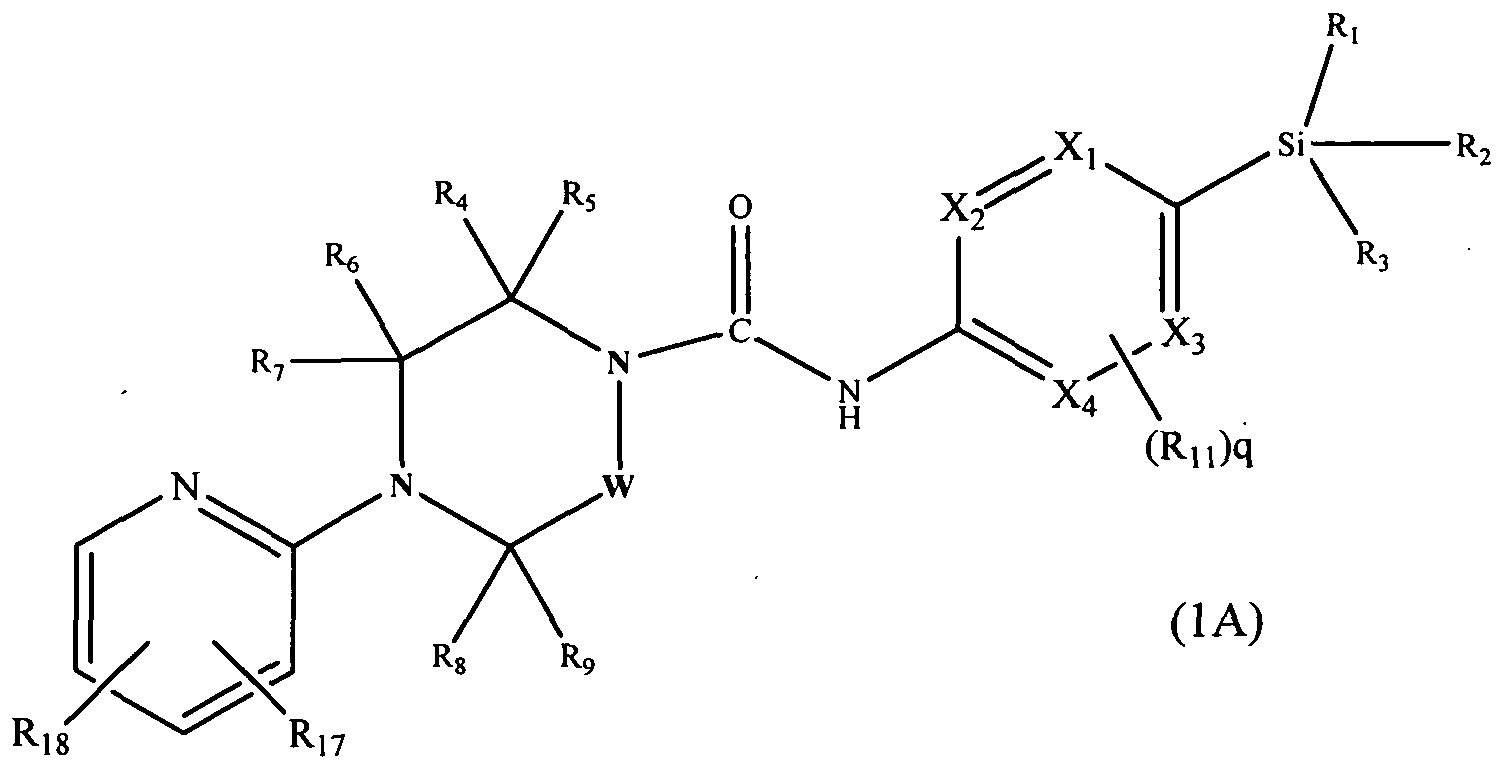
[0081]wherein Xi, X2, X3, X4, W, R1, R2, R3, R4, R5, RO, R7, Rs, R9, Rn and q are defined as herein, wherein R17 and R18 may be present or absent and independently represent halogen, haloCi-6alkyl,
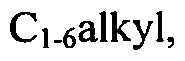 -Q2-S(O)1nRi2 - CN, -Q2-CO2Rn, -Q2-CONR,2R13> hydroxyC1-6alkyl, -Q2-NR12R13, -Q2-ORn or -Q2-SO2NR12R]3, or a pharmaceutically acceptable salt or ester thereof, and wherein Q2, Rn, R]3 and m are as defined herein.
-Q2-S(O)1nRi2 - CN, -Q2-CO2Rn, -Q2-CONR,2R13> hydroxyC1-6alkyl, -Q2-NR12R13, -Q2-ORn or -Q2-SO2NR12R]3, or a pharmaceutically acceptable salt or ester thereof, and wherein Q2, Rn, R]3 and m are as defined herein.
[0082]In an embodiment Rn and R|g independently represent halogen (e.g. chloro, or fluoro), haloC1-6alkyl (e.g. trifluoromethyl), CI-6alkyl (e.g. methyl), -Q2-S(O)1nRi2 (e.g. methylsulphonyl), -CN, -Q2-CO2R]2 (e.g. carboxy, ethoxycarbonyl, methoxycarbonyl, carboxymethyl, methoxycarbonylmethyl), -Q2-CONR12R13 (e.g. carbamoyl, dimethylcarbamoyl or pyrrolidine- 1 -ylcarbonyl), hydroxyC1-6alkyl (e.g. hydroxymethyl, hydroxyisopropyl or hydroxyethyl), -Q2-NR12R13 (e.g. dimethylaminomethyl), -Q2-OR12 (e.g. methoxymethoxymethyl, methoxymethyl), or -Q2-SO2NRnR13 (e.g. dimethylaminosulphonyl).
[0083]In another embodiment R17 is attached at position 3 of the pyridin-2-yl ring.
[0084]In a further embodiment R17 is attached at position 3 of the pyridin-2-yl ring and represents halogen (e.g. fluoro, chloro). In a further embodiment R]7 is attached at position 3 of the pyridin-2-yl ring and represents chloro.
[0085]In one embodiment R]g is attached at position 5 of the pyridin-2-yl ring.
[0086]In a further embodiment R^ is attached at position 5 of the pyridin-2-yl ring and represents -Q2-CO2Rn (e.g. carboxy, ethoxycarbonyl, methoxycarbonyl, carboxymethyl, methoxycarbonylmethyl), -Q2-CONRi2Ri3 (e.g. carbamoyl, dimethylcarbamoyl or pyrrolidine- 1 -ylcarbonyl), -Q2-SO2NRnR)3 (e.g. dimethylaminosulphonyl), or -Q2-ORn, (e.g. methoxymethoxymethyl, methoxymethyl).
[0087]A further subclass of compounds according to the invention is represented by the compounds of formula 1 (B)>
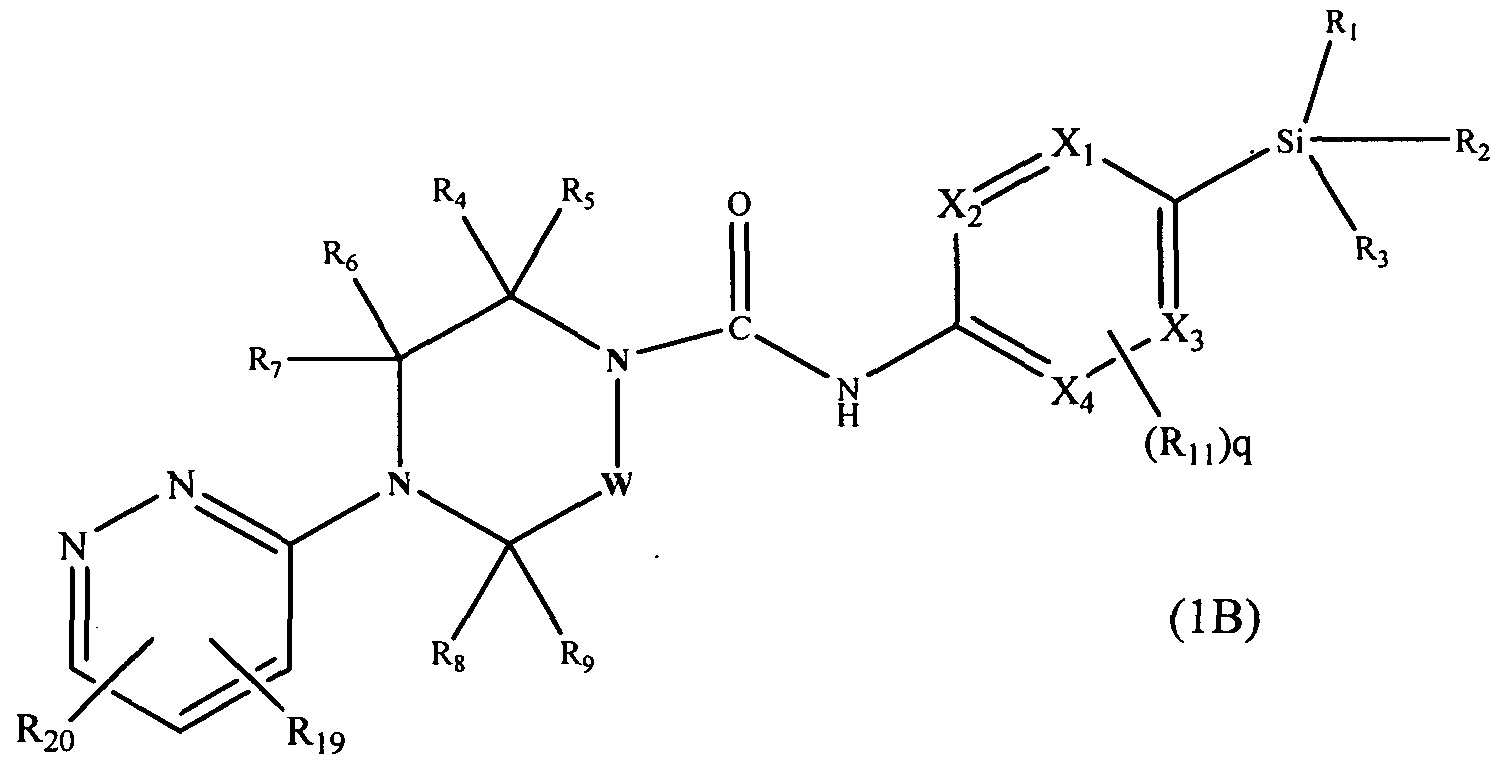
[0088]wherein X1, X2, X3, X4, W, R1, R2, R3, R4, R5, Re, R7, Rs, R9, Rn and q are defined as herein, wherein R19 may be present or absent and represents C^alkyl (e.g. methyl, ethyl), and wherein R20 may be present or absent and represents halogen.
[0089]In an embodiment R19 is attached at position 4 of the pyridazin-3-yl ring and represents Ci-6alkyl (e.g.methyl).
[0090]In a further embodiment R20 represents halogen (e.g. chloro) and is attached at position 6 of the pyridazin-3- yl ring.
[0091]A further subclass of compounds according to the invention is represented by the compounds of formula l(C):-
[0092]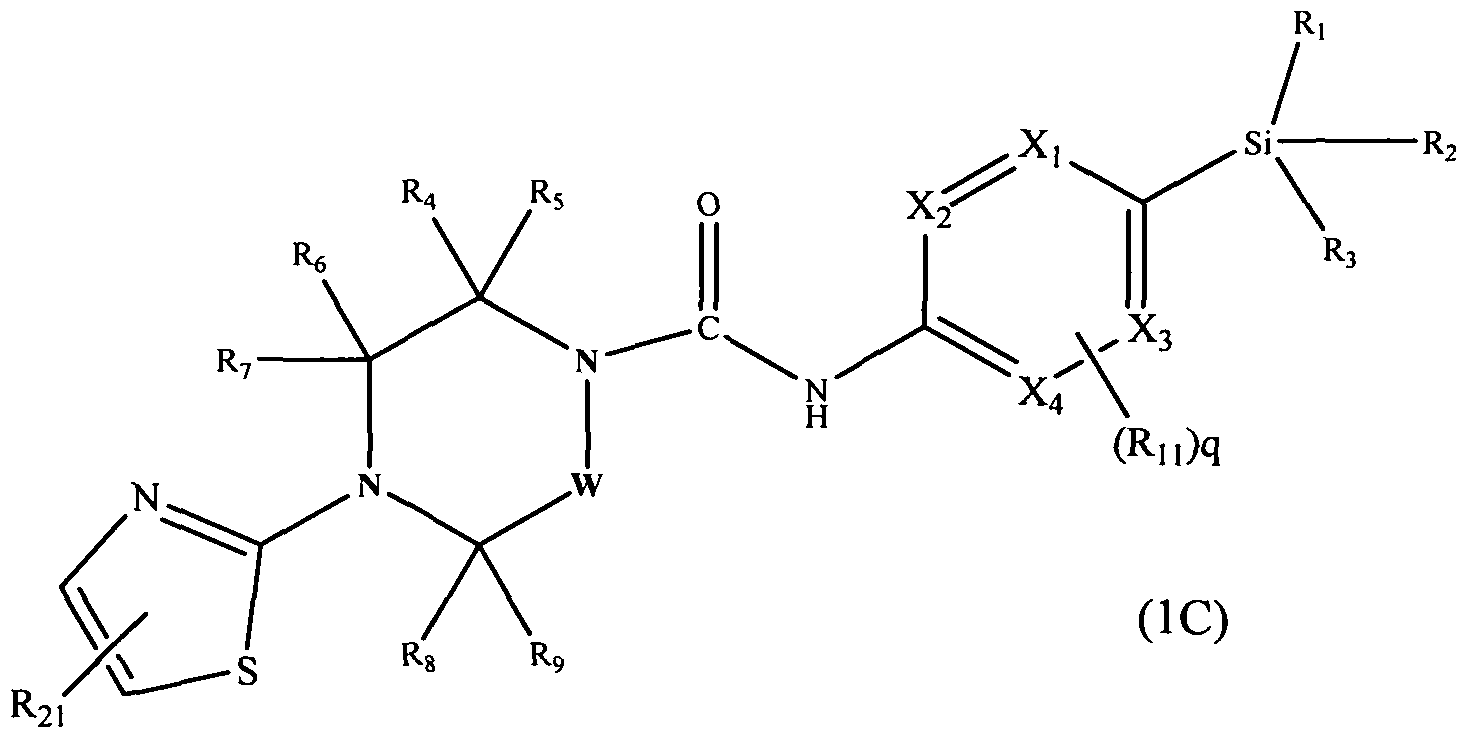
[0093]wherein X1, X2, X3, X4, W, R1, R2, R3, R4, R5, R6, R7, R8, R9, Rn and q are as defined herein, and wherein R2] may be present or absent and represents -Q2-CO2Rj2 (e.g. carboxy, methoxycarbonyl), hydroxyC^alkyl (e.g. hydroxymethyl), or -Q2-CONRi2Ru (e.g. methylcarbamoyl dimethylcarbamoyl, pyrrolidine- 1-ylcarbonyl).
[0094]In an embodiment R21 is attached at position 4 of the thiazol-2-yl ring. In a further embodiment R21 is attached at position 5 of the thiazol-2-yl ring.
A further subclass of compounds according to the invention is represented by the compounds of formula l(D):-
[0095]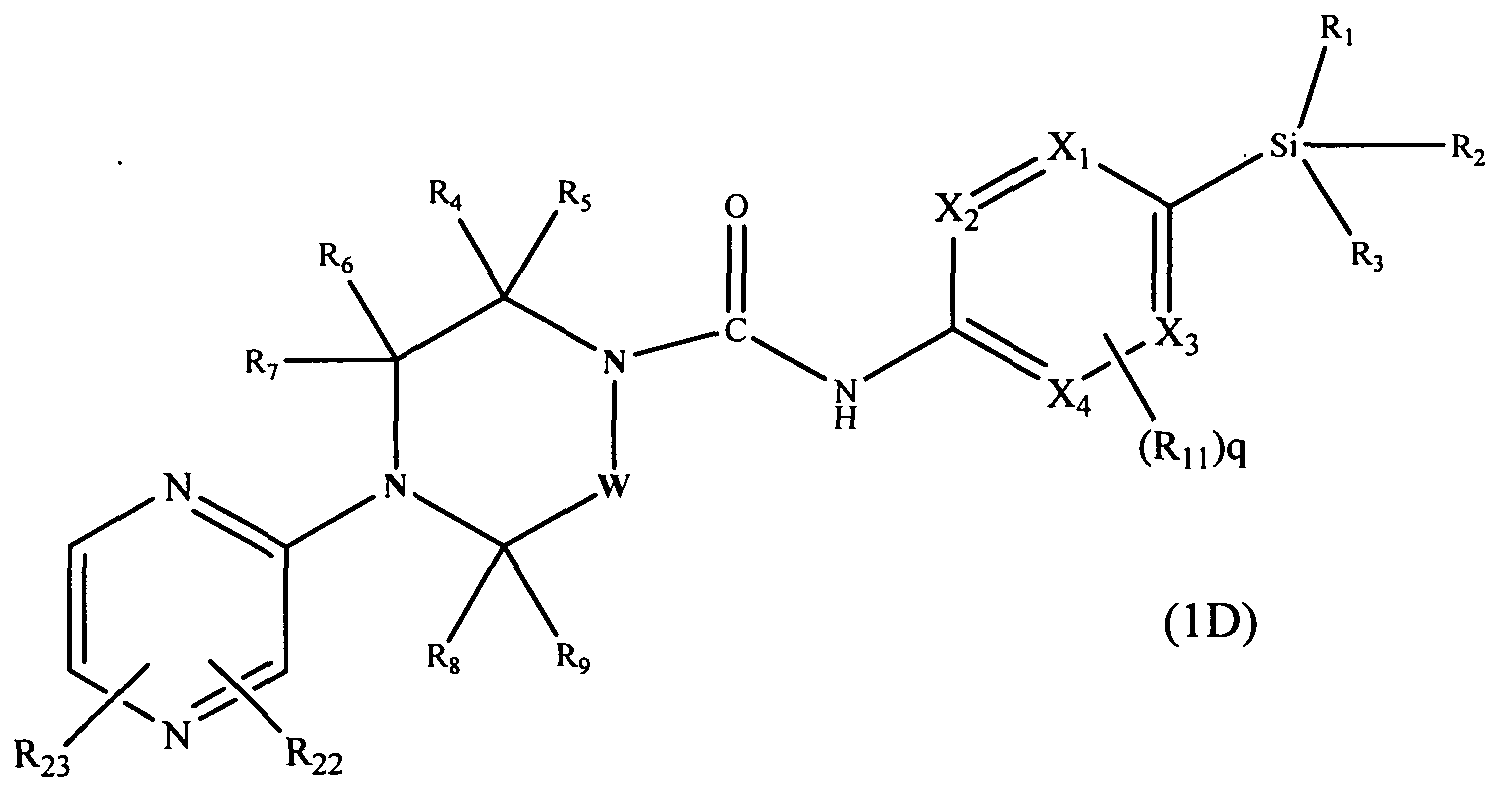
[0096]wherein Xi, X2, X3, X4, W, Ri, R2, R3, R4, R5, Re, R7, Rs, R9. Ru and q are defined as herein, wherein R22 and R23 may be present or absent and independently represent Ci^alkyl (e.g. methyl, ethyl), -Q2-CO2Rn (e.g. methoxycarbonyl, carbonyl), or -Q2-CONRi2Rn (e.g. aminocarbonyl, methylaminocarbonyl).
[0097]In an embodiment R22 and R23 independently represent C)-6alkyI (e.g. methyl), Q2-CO2R12 (e.g. methoxycarbonyl), or -Q2-CONRi2Ri3 (e-g- aminocarbonyl)
[0098]In a further embodiment R23 is absent and R22 represents C^alkyl (e.g. methyl) and may be attached at the 3 position of the pyrazin-2-yl ring.
[0099]In a further embodiment R22 represents Ci^alkyl (e.g. methyl) and R23 represents aminocarbonyl, wherein R22 may be attached at the 3 position of the pyrazin-2-yl ring.
[0100]A further subclass of compounds according to the invention is represented by the compounds of formula l(E):-
[0101]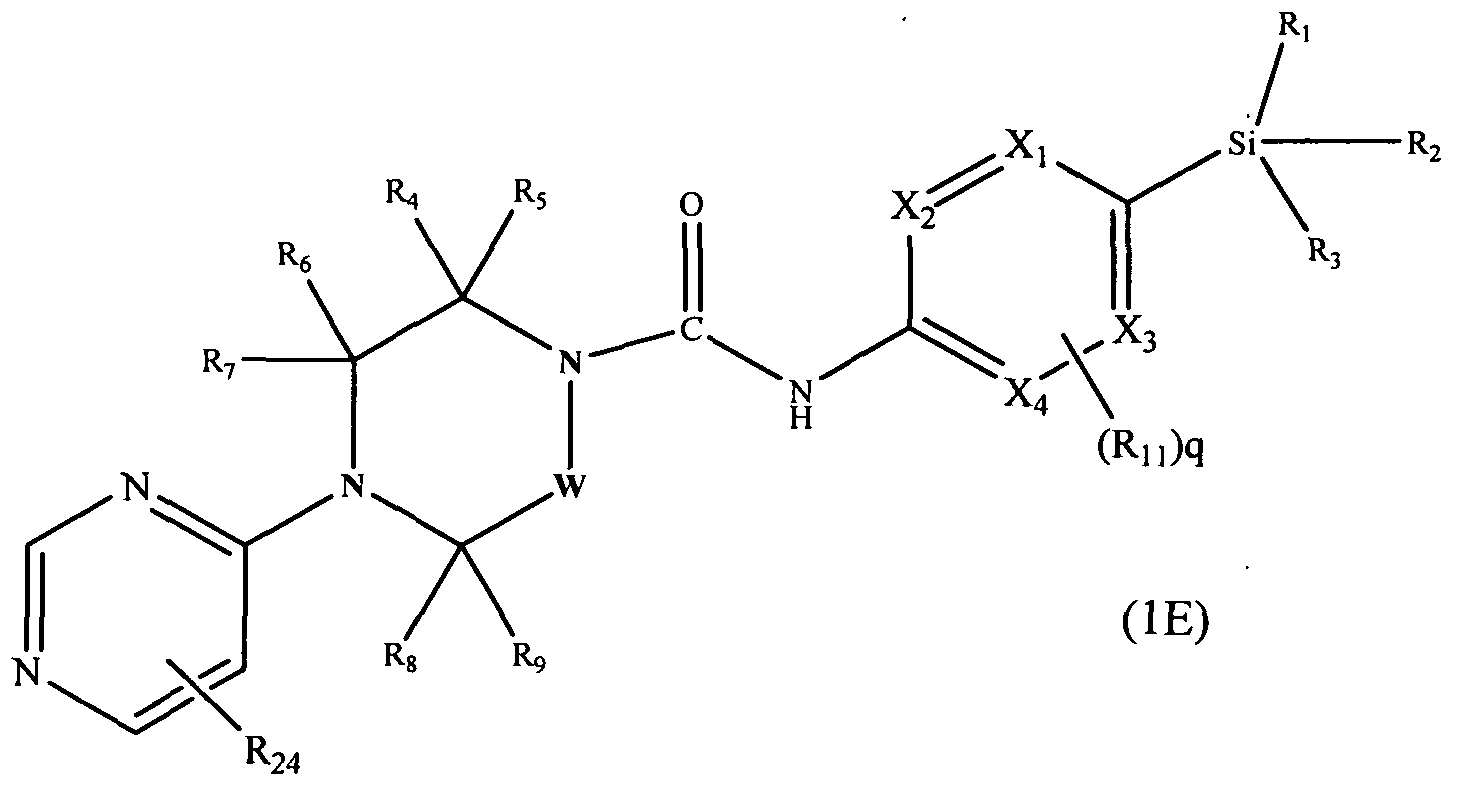
[0102]wherein Xi, X2, X3, X4, W, Ri, R2, R3, R4, R5, Re, R7, Rs, R9, Rn and q are defined as herein, wherein R^ may be present or absent and represents Ci-6alkyl (e.g. methyl, ethyl), or halogen (e.g. chloro, fluoro).
[0103]In one embodiment R24 is attached at position 5 of the pyrimidin-4-yl ring.
[0104]In a further embodiment R24 represents halogen (e.g. fluoro), or C^aHcyl (e.g. methyl).
[0105]A further subclass of compounds according to the invention is represented by the compounds of formula l(F):-
[0106]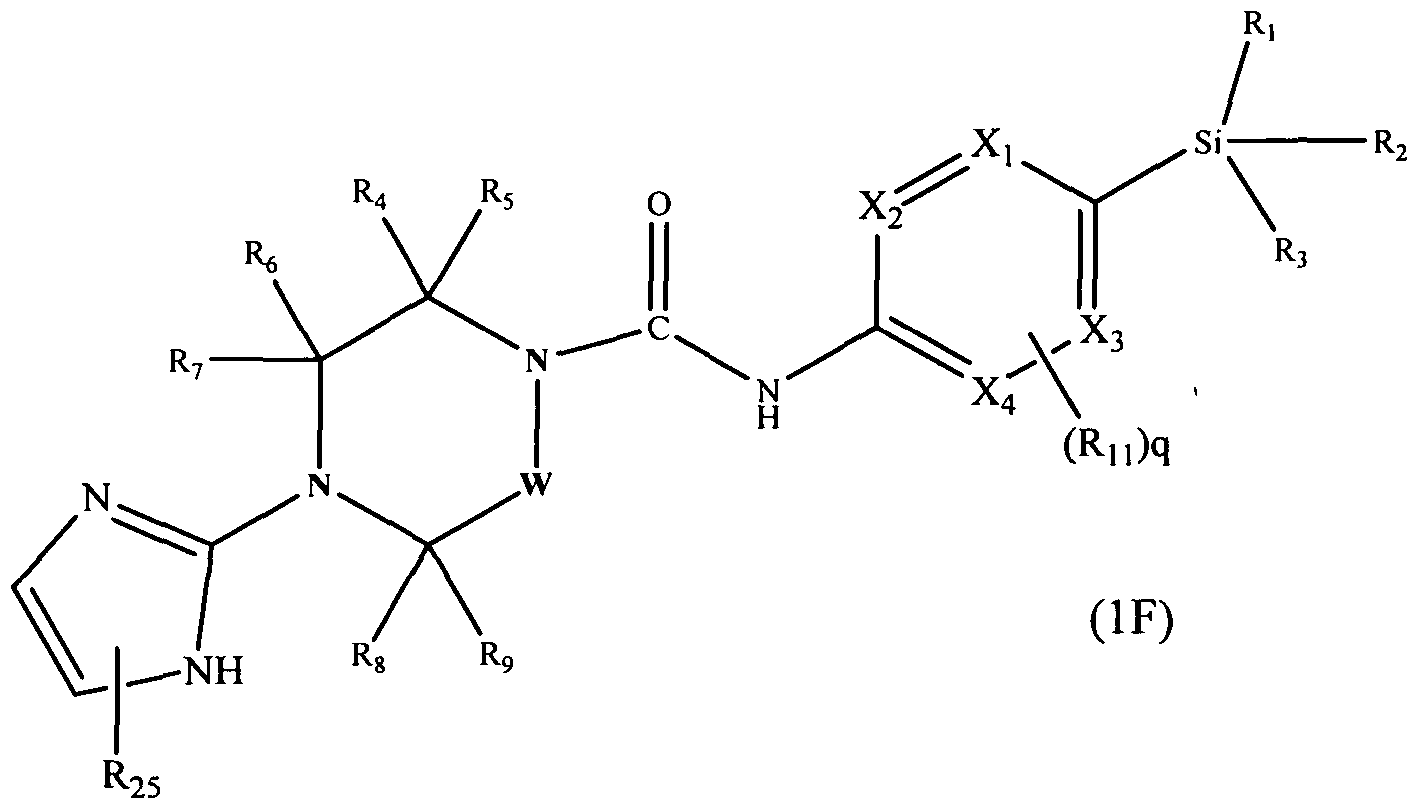
[0107]wherein Xi, X2, X3, X4, W, Rj, R2, R3, R4, R5, Rs, R7, Rs, R9, Rn and q are defined as herein, wherein R2s may be present or absent and represents Ci^alkyl (e.g. methyl, ethyl), or halogen (e.g. chloro, fluoro).
[0108]In one embodiment R25 is attached to the N at position 1 of the imidazol-2-yl ring. In a further embodiment R25 represents methyl.
In particular compounds of formulae l(A) to l(F), the pyridyl, pyridazinyl, thiazolyl, pyrazinyl, pyrimidyl and imidazolyl groups, respectively representing R10, may be selected from the 'Specific examples of R)0' given above.
[0109]In accordance with a second aspect of the invention, there is provided a pharmaceutical composition comprising a compound according to the first aspect of the invention, together with one or more pharmaceutically acceptable excipients.
[0110]Pharmaceutical compositions of this invention comprise any of the compounds of the first aspect of the present invention, or pharmaceutically acceptable salts and esters thereof, with any pharmaceutically acceptable carrier, adjuvant or vehicle. Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of this invention are those conventionally employed in the field of pharmaceutical formulation, and include, but are not limited to, sugars, sugar alcohols, starches, ion exchangers, alumina, aluminium stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycerine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulphate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene- polyoxypropylene-block polymers, polyethylene glycol and wool fat.
[0111]The pharmaceutical compositions of this invention may be administered orally, parenterally, by inhalation spray, rectally, dermally, intra-vesically, nasally, buccally, vaginally or via an implanted reservoir. Oral administration is preferred. The pharmaceutical compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers, adjuvants or vehicles. The term parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intra-articular, intrasynovial, intrasternal, intrathecal, intralesional and intracranial injection or infusion techniques.
[0112]The pharmaceutical compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant such as that described in Ph. HeIv, or a similar alcohol.
The pharmaceutical compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, powders, granules, and aqueous suspensions and solutions. These dosage forms are prepared according to techniques well-known in the art of pharmaceutical formulation. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavouring and/or colouring agents may be added.
[0113]The pharmaceutical compositions of this invention may also be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
[0114]The pharmaceutical compositions of this invention may be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilising or dispersing agents known in the art.
[0115]The compounds of the present invention may be administered to the subject (preferably a mammal, more preferably a human) in a dose of around 1 to around 20,000 μg/kg body weight per dose, depending on the condition to be treated or prevented, and the characteristics of the subject being administered with the compound. In many instances, the dose may be around 1 to around 1500 μg/kg body weight per dose. The dosing regimen for a given compound could readily be determined by the skilled person having access to this disclosure. For instance, the above dose can be given to the subject one to three times per day.
[0116]The pharmaceutical compositions of the present invention may contain, in addition to the compounds of the first aspect of the invention, one or more additional active pharmaceutical ingredients known to be efficacious in the treatment or prevention of the conditions indicated herein, or in the treatment of comorbidities of those conditions.
[0117]In a third aspect, the present invention provides a compound according to the first aspect of the invention, or a composition according to the second aspect, for use in therapy.
[0118]In a fourth aspect, the invention provides a compound according to the first aspect of the invention, or a composition according to the second aspect, for use in the treatment or prevention of a condition of which the development or symptoms are linked to TrpVl receptor activity.
[0119]A number of conditions of which the development or symptoms are linked to TrpVl receptor activity are known to the skilled person.
In a fifth aspect, the invention also provides a method of treatment or prevention of a condition of which the development or symptoms are linked to TrpVl receptor activity, the method comprising the administration, to a subject in need of such treatment or prevention, of a therapeutically effective amount of a compound according to the first aspect of the invention, or a composition according to the second aspect.
[0120]In a sixth aspect, the invention also provides the use of a compound according to the first aspect in the preparation of a medicament for the treatment or prevention of a condition of which the development or symptoms are linked to TrpVl receptor activity.
[0121]In particular embodiments of the compound according to the fourth aspect, the method according to the fifth aspect, or the use according to the sixth aspect, the condition is pain, a urological disorder or urinary dysfunction, an inflammatory disorder or a disorder involving sensory nerve function.
[0122]In certain embodiments, the condition to be treated or prevented may be selected from chronic inflammatory pain, musculo-joint pain, (osteo)arthritic pain, rheumatic pain, post-operative pain, dental pain (such as following third molar extraction), post-mastectomy pain, neuropathic pain, pain associated with nerve damage, diabetes-induced neuropathy, pain associated with substance abuse (such as alcohol or narcotic), burn pain (such as sunburn, UV burn, radiant heat burn, chemical burn), glossodynia, cold induced pain (such as frostbite), cold allodynia, pain associated with exposure to TrpVl agonists or animal bites and stings, central pain including fibromyalgia, pain associated with infection (such as post-herpetic neuralgia), HIV- induced neuropathy, chemotherapy induced neuropathy, cancer induced pain (including bone cancer), pain associated with amputations or 'phantom limb pain', nerve entrapment or brachial plexus avulsions, lower back pain, sciatica, ankylosing spondylitis, complex regional pain syndrome, reflex sympathetic dystrophy, chronic nerve injury, headache (such as migraine, cluster headache and tension headache), temporomandibular pain, maxilary sinus pain, ear pain, trigeminal neuralgia, pain associated with stroke, multiple sclerosis, gout, scar pain, post-polio syndrome, visceral pain (such as heart pain, muscle pain, eye pain, orofacial pain, abdominal pain, gynaecological pain), hemorrhoids, gastrointestinal disorders including inflammatory bowel disorders (such as ulcerative colitis, ileitis, Crohns Disease, Barrett's syndrome), inflammatory bowel syndrome, stomach cramps, bloating, diarrhoea, pancreatitis, bladder disorders (such as urinary dysfunctions, bladder overactivity, urinary incontinence, urge incontinence, stress incontinence, mixed stress/urge incontinence, neurogenic incontinence, bladder detrusor hyperreflexia, detrusor instability, urinary outlet obstruction, nephritis, sensory urgency, motor urgency, nocturia, painful bladder syndrome, bladder associated visceral pain, cystitis, interstitial cystitis and vulvodynia), respiratory disorders (such as chronic obstructive pulmonary or airway disorder (COPD or COAD), adult respiratory distress syndrome (ARDS), chronic bronchitis, pneumoconiosis, rhinitis, including allergic and non-allergic rhinitis, asthma, cough including idiopathic cough and cough associated with diseases such as asthma, cystic fibrosis, cancer) and gastrointestinal disturbances such as reflux, inflammatory skin disorders (such as psoriasis, eczema, itch of non-specific origin, contact dermatitis and hypersensitivity), depression, anxiety, stroke, myocardial infarction, inflammatory eye disorders (such as uveitis), traumatic brain injury, spinal cord injury, neurodegenerative diseases (such as Parkinson's disease, Alzheimer's disease), renal disorders, obesity,
schizophrenia, epilepsy, sleeping disorders, cancer (such as bone cancer) and elevated blood pressure, diabetes including type I diabetes and type II diabetes, and arthritis.
[0123]In particular embodiments, the condition to be treated or prevented may be selected from chronic inflammatory pain, (osteo)arthritic pain, dental pain, neuropathic pain, HIV-induced neuropathy, chemotherapy induced neuropathy, cancer induced pain, pain associated with stroke, visceral pain, gastrointestinal disorders, pancreatitis, bladder disorders, respiratory disorders, cough including idiopathic cough, and diabetes.
[0124]In some embodiments, the condition may be selected from urological disorders and pain.
[0125]In the following process description, the symbols Ri, R2, R3, R4, R5, Re, R7, Rs, R9, Rio, Rn, W, X1, X2, X3 and X4 when used in the formulae depicted are to be understood to represent those groups as described above in relation to formula (1) unless otherwise indicated. During any of the synthetic sequences it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. The methods of addition and removal of such protecting groups are those which would conventionally be used in relation to the particular molecule-type or group being protected, for example the methods described in standard works of reference in synthetic methodology, such as Kocienski (2004) Protecting Groups. 4th Edn. Georg Thieme Verlag. In some instances, deprotection may be the final step in the synthesis of a compound of formula (1) and the processes according to the invention described hereinafter are to be understood to extend to such removal of protecting groups.
[0126]The following processes together with the intermediates are provided as further aspects of the invention.
[0127]Thus in another aspect of the invention, the compounds of formula (1) may be prepared by a process which comprises reacting a compound of formula (i) with a compound of formula (ii):
[0128]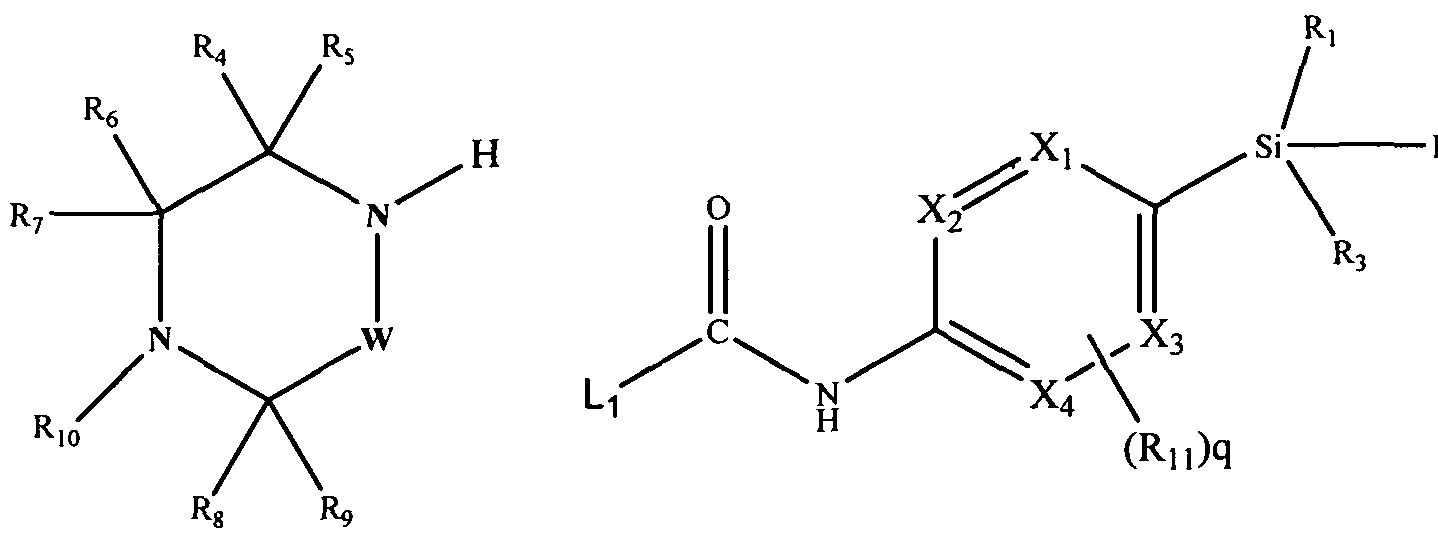
[0129](i) (ϋ) wherein Ri, R2, R3, R4, R5, R^, R7, R8, R9, Ri0, Rn, W, X1, X2, X3 and X4 are as defined above, and L) represents a suitable leaving group.
[0130]The leaving group L1 may be phenoxy.
[0131]The reaction may conveniently be effected at ambient temperature in a suitable solvent such as a cyclic ether e.g. tetrahydrofuran in the presence of an organic amine such as l,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
Alternatively the reaction may be effected at elevated temperature in a suitable solvent e.g. a cyclic ether such as tetrahydrofuran or a lower alcohol e.g. ethanol using standard equipment or a microwave reactor. The reaction may also be performed in the presence of an organic amine such as triethylamiπe, typically in the presence of a suitable catalyst such as 4-dimethylaminopyridine (DMAP).
[0132]Where they are not commercially available the starting materials of formula (i) and (ii) may be prepared using methods analogous to those as described in the Examples hereinafter or using standard methodology known to the person skilled in the art.
[0133]For example, the starting material of formula (i) may be prepared by a process which comprises reacting a compound of formula (iii) with a compound of formula R10-L2:
[0134]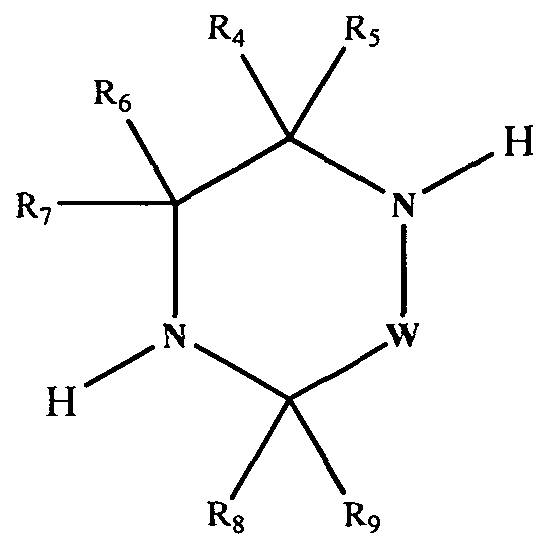
[0135](iii) wherein R4, R5, R^, R7, Rs, R9, Rio and W are as defined above, and L2 represents a suitable leaving group attached to a carbon atom adjacent to the ring nitrogen of a nitrogen heteroaryl group Ri0.
[0136]The leaving group L2 is typically a halogen atom, e.g. chloro, fluoro or bromo.
[0137]The reaction may be effected at ambient or elevated temperature in a suitable solvent e.g. DMSO, acetonitrile or a lower alcohol e.g. propan-2-ol using standard equipment or a microwave reactor. The reaction may be performed in the presence of an organic base such as triethylamine.
[0138]Alternatively when R]0 is an aryl group, or a heteroaryl group having a leaving group attached to a carbon atom not adjacent to a ring nitrogen, the reaction may be effected by treatment with a palladium catalyst such as Pd(OAc)2 and BINAP, NaO1Bu at elevated temperature, e.g. microwave in a suitable solvent such as toluene.
[0139]The intermediates of formula (iii) and RjO-L2 where they are not commercially available may be prepared using standard methods known to those skilled in the art or methods analogous to those described in the Examples.
[0140]It will be understood that any compound of formula (1) initially obtained from any of the above processes may be further elaborated into a further compound of formula (1) using methods known in the art. By way of example a compound of formula (1) wherein Rj0 is substituted with an ester such as -Q2-CO2CH3 Or -Q2- CO2CH2CH3, where Q2 is as herein defined, may be hydrolysed to the corresponding carboxylic acid -Q2- CO2H under basic conditions e.g. using an inorganic base such as NaOH or KOH. The resulting carboxylic
acid may be further reacted e.g. with an amine to give the corresponding amide or e.g. with a reducing agent, such as LiBH4 to give the corresponding methylalcohol. Alternatively a compound of formula (1) wherein Rio is substituted with an ester such as -Q2-CO2CH3 or -Q2-CO2CH2CH3, where Q2 is as herein defined, may be reduced to the corresponding methylalcohol using a reducing agent such as LiAlH4 or reacted with ammonium chloride in the presence of 'butylammonium bromide to yield the corresponding amide -Q2- CONH2. By way of further example a compound of formula (1) wherein R]0 is substituted by an amine of formula -Q2-NR12R13 may be prepared from the corresponding alcohol -Q2-OH by initial conversion into a suitable leaving group, e.g. methanesulfonyl formed by reaction of the alcohol with e.g. methanesulfonyl chloride in the presence of an organic base such as triethylamine, followed by displacement with an amine of formula HNRi2Ru in the presence of an organic amine such as triethylamine.
[0141]By way of a further example a compound of formula (1) may be elaborated into an N-oxide of formula (1) using for example an oxidizing agent such as mCPBA in the presence of sodium bicarbonate in an appropriate solvent e.g. a halogenated hydrocarbon such as dichloromethane. Such N-oxide is also within the scope of the compound according to the present invention.
[0142]In another example a compound of formula (1) wherein R10 is substituted with a halogen atom, e.g. chloro, may be hydrogenated in the presence of a palladium catalyst to give a further compound of formula (1) in which the halogen atom has been removed.
[0143]All novel intermediates form a further aspect of the invention.
[0144]The invention will now be described in more detail by way of example only.
[0145]1. Synthetic Methodologies
[0146]The methods used for synthesis of the compounds of the invention are illustrated by the general schemes below and the preparative examples that follow. All compounds and intermediates were characterised at least by liquid chromatography-mass spectroscopy (LCMS). The starting materials and reagents used in preparing these compounds are available from commercial suppliers. These general schemes are merely illustrative of methods by which the compounds of this invention can be synthesised, and various modifications to these schemes can be made and will be suggested to one skilled in the art having referred to this disclosure.
[0147]Nuclear magnetic resonance (NMR) spectra were recorded at 400MHz; the chemical shifts (δ) are reported in parts per million. Spectra were recorded using a Bruker 400 Avance instrument fitted with a 5mm BBFO probe (DUL probe prior to October 2008). Instrument control was by Bruker TopSpin 2.1 software.
[0148]Purity was assessed using UPLC with UV (photodiode array) detection over a wide range of wavelengths, normally 220-450nm, using a Waters Acquity UPLC system equipped with Acquity UPLC BEH or HSS Cl 8 columns (2.1mm id x 50mm long) operated at 50 or 6O0C. Mobile phases typically consisted of acetonitrile or methanol mixed with water containing either 0.05% formic acid or 0.025% ammonia. Mass spectra were recorded with a Waters SQD single quadrupole mass spectrometer using atmospheric pressure ionisation.
Compounds were purified using normal phase chromatography on silica or alumina, or by reverse phase chromatographic methods, using Biotage or Isolute KPNH Cartridge, SCX cartridge and SCX-2 solid phase extraction cartridges.
[0149]Preparative HPLC was performed using an Agilent Technologies 1100 Series system typically using Waters 19mm id x 100mm long Cl 8 columns such as XBridge or SunFire 5μm materials at room temperature. Mobile phases typically consisted of acetonitrile or methanol mixed with water containing either 0.1 % formic acid or 0.1% ammonia.
[0150]Room temperature in the following schemes means the temperature ranging from 2O0C to 250C.
[0152]DBU - l,8-Diazabicyclo[5.4.0]undec-7-ene
[0153]THF - Tetrahydrofuran
[0154]BINAP - 2,2'-bis(diphenylphosphino )-l,l'-binaphthyl
[0155]TEA - Triethylamine
[0156]DMAP - 4-Dimethylaminopyridine
[0157]DMSO - Dimethyl sulfoxide
[0159]EDC - l-ethyl-3-(3-dimethylaminopropyl) carbodiimide
[0160]Pd(OAc)2 - Palladium acetate
[0161]DCM - Dichloromethane
[0162]MsCl - Mesyl chloride rt - room temperature
[0164]MeOD - Deuterated methanol
[0165]Boc - Butoxycarbonyl mCPBA - meta-chloroperoxybenzoic acid
[0166]MP-carbonate - Macroporous triethylammonium methylpolystyrene carbonate
[0167]HOAt - l-Hydroxy-7-Azabenzotriazole
[0168]Schemes 1.1 - 1.17 serve to illustrate the methodologies that may be used to synthesize the exemplified compounds of formula (1)
[0169]Synthesis of Intermediate 1
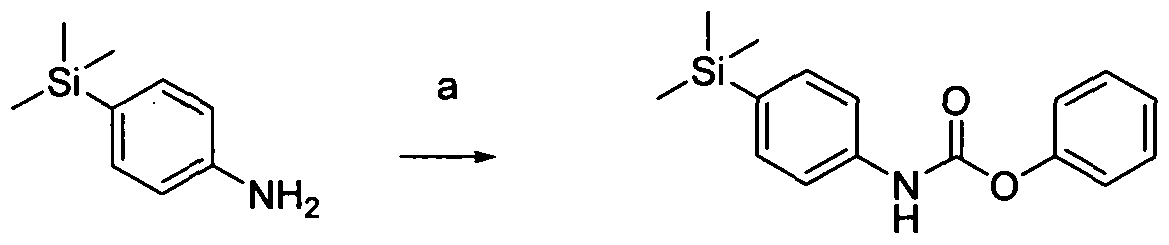
[0171]Reagents and conditions: a) phenylchloroformate, TEA, DCM, rt 16hr
[0172]Phenyl-4-(trimethylsilyl)phenylcarbamate Intermediate 1. A solution of 4-trimethylsilylaniline [Benkeser, Robert A.; Journal of the American Chemical Society 1952, V74, P253-4] (30mmol), phenylchloroformate (33.3mmol) and TEA (104mmol) in DCM (250ml) was stirred at room temperature for 16 hrs. The reaction mixture was washed with citric acid followed by brine and the organic was separated, dried (phase sep.) and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (20-50% DCM in petroleum ether) yielding the product (21.9mmol).1H NMR (400 MHz, DMSOd6) δ 7.16 - 7.35 (m, 6H), 6.91 - 7.16 (m, 3H), 0.00 (s, 9H).
[0174]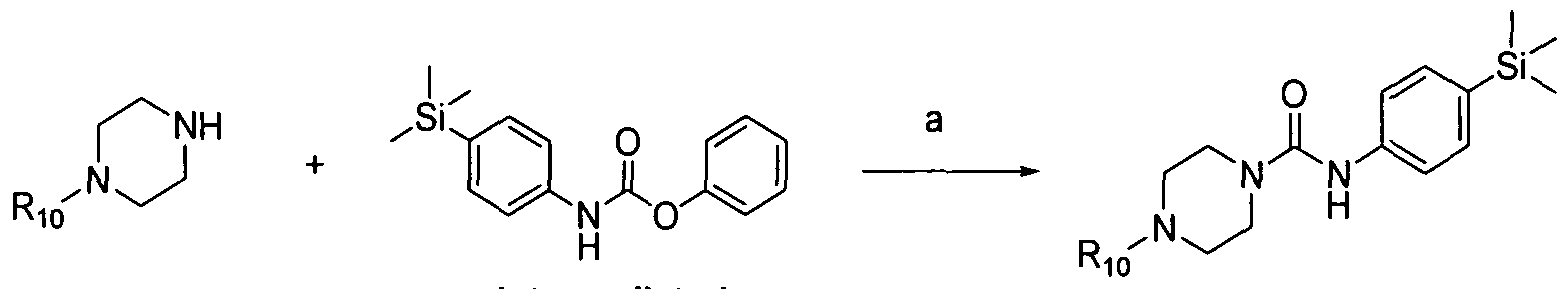
[0176]Reagents and conditions: a) DBU, THF rt 3hrs
[0177]wherein Rio is an appropriately substituted aryl or heteroaryl group, which may bear optional substituents described by or which can be modified to the optional substituents described for R!0 according to the first aspect of the invention.
[0179]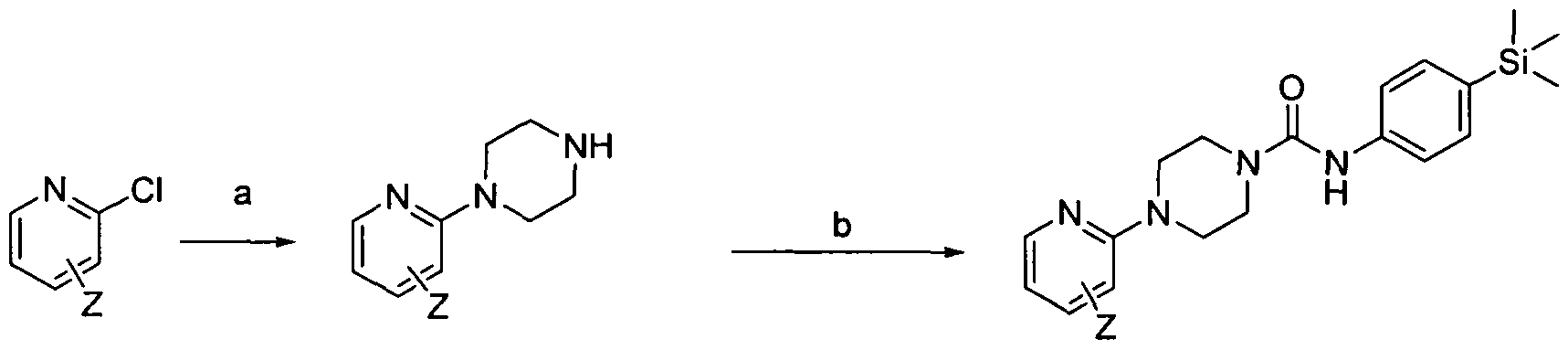
[0180]Reagents and conditions: a) melt in microwave 110°C 30 min, b) Intermediate 1 , THF rt 3hrs wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention. Step (a) of Scheme 2 employs a piperazine, which may be protected (e.g. 1 -Boc-piperazine).
[0181]1.3 Scheme 3
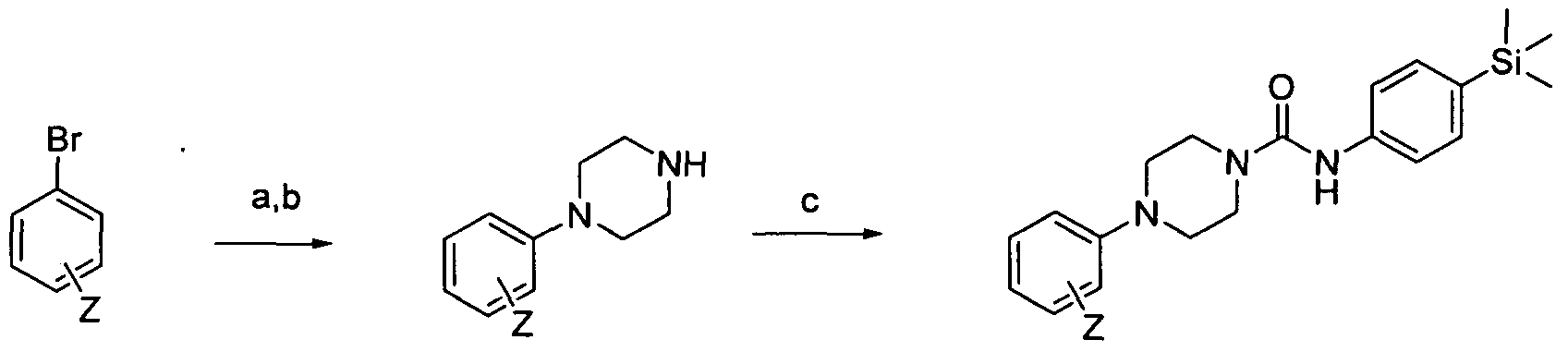
[0182]Reagents and conditions: a) 1-Boc-piperazine, Pd(OAc)2, BINAP, NaO4Bu, Toluene microwave 120°C 30 min b) HCI-dioxane rt 18hrs c) Intermediate 1 , DBU, THF rt 3hrs
[0183]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0185]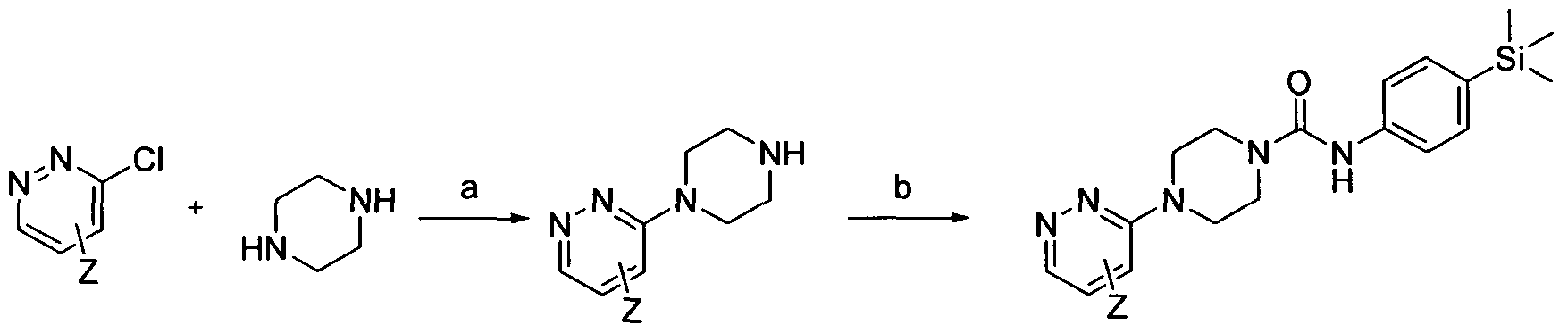
[0186]Reagents and conditions: a) TEA, DMSO, 100°C 18 hrs b) Intermediate 1 , EtOH microwave 30 min 100 °C
[0187]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0189]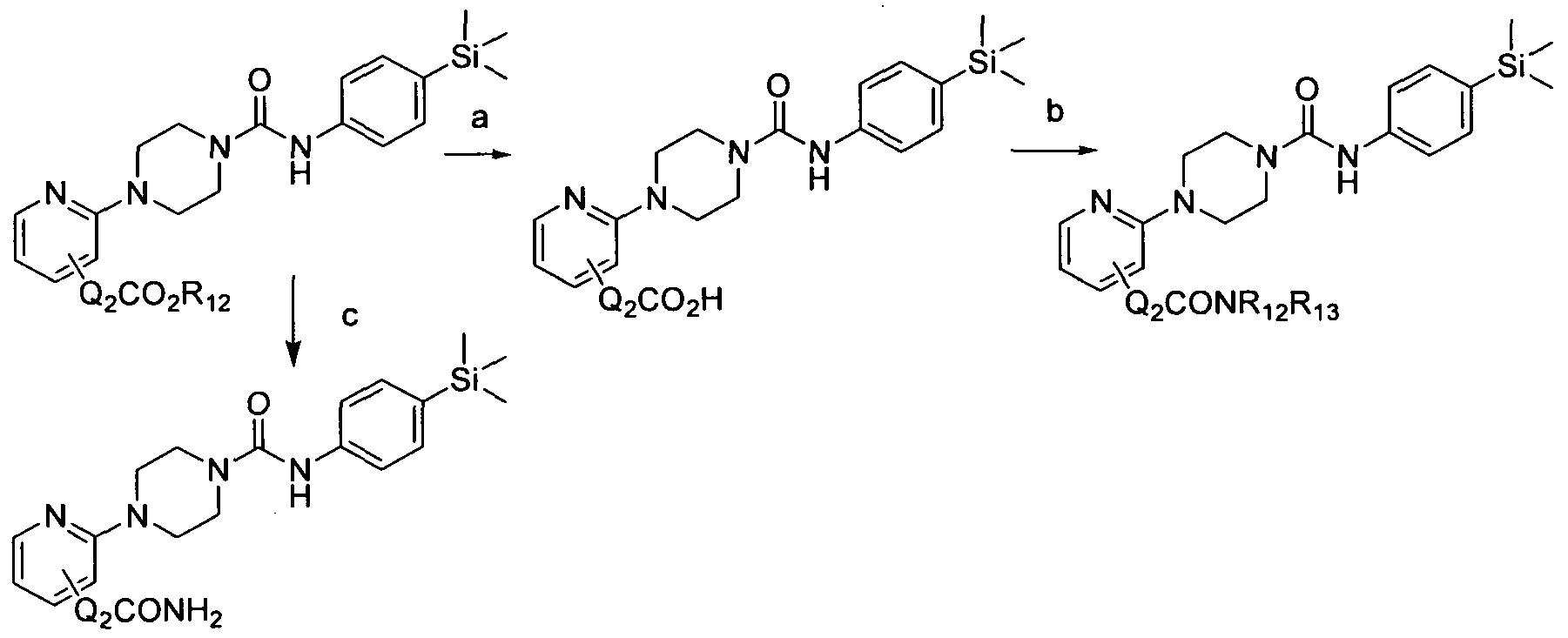
[0190]Reagents and conditions: a) KOH, THF, 60 hrs b) EDC, R12R13NH, TEA, DCM, rt, 3 days c) NH4CI,1BuNH3Br, NH4OH(aq)
[0191]wherein Q2 and Ri2 and Rn are as defined herein. 1.6 Scheme 6
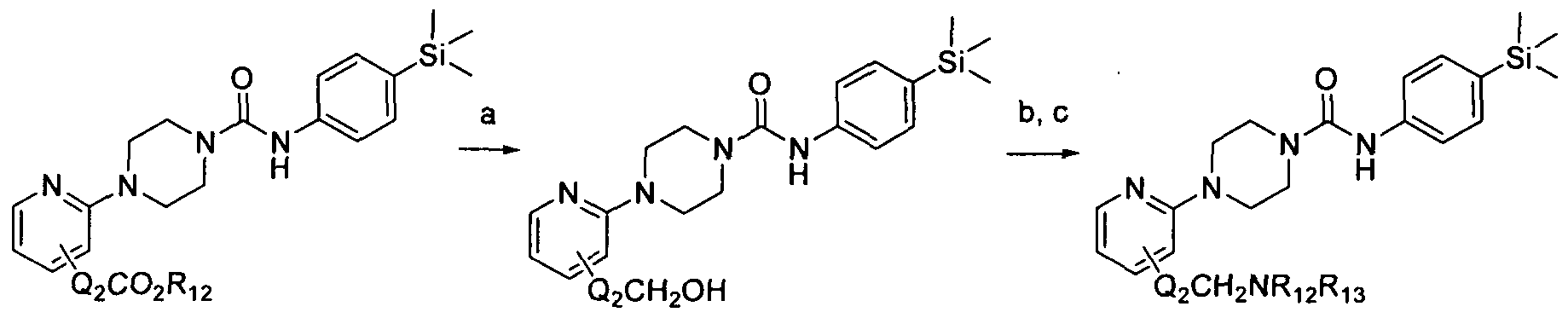
[0192]Reagents and conditions: a) LiBH4, EtOH, rt, 24hrs b) MsCI, TEA, DCM, rt, 2hrs c) R12R13NH, TEA, DCM, rt, 18hrs wherein Q2 and R12 and R13 are as defined herein. 1.7 Scheme 7
[0193]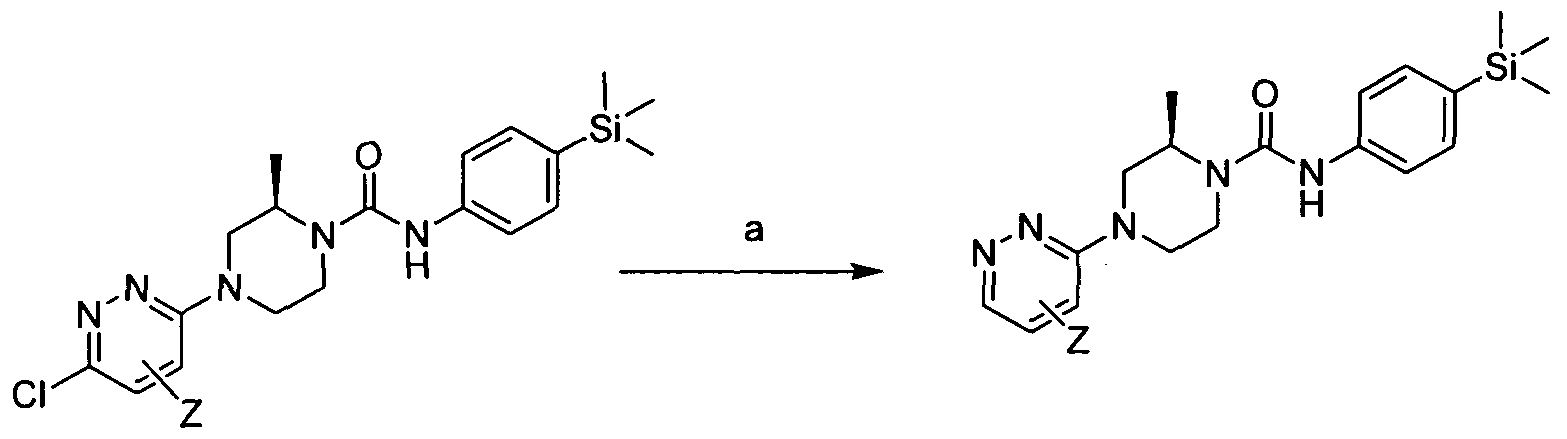
[0194]Reagents and conditions: a) EtOH, H2(g), Pd/C 30 min 500C wherewhe rein Z is a group that is described by, or which can be modified to, the optional substituents described for R)o according to the first aspect of the invention.
[0196]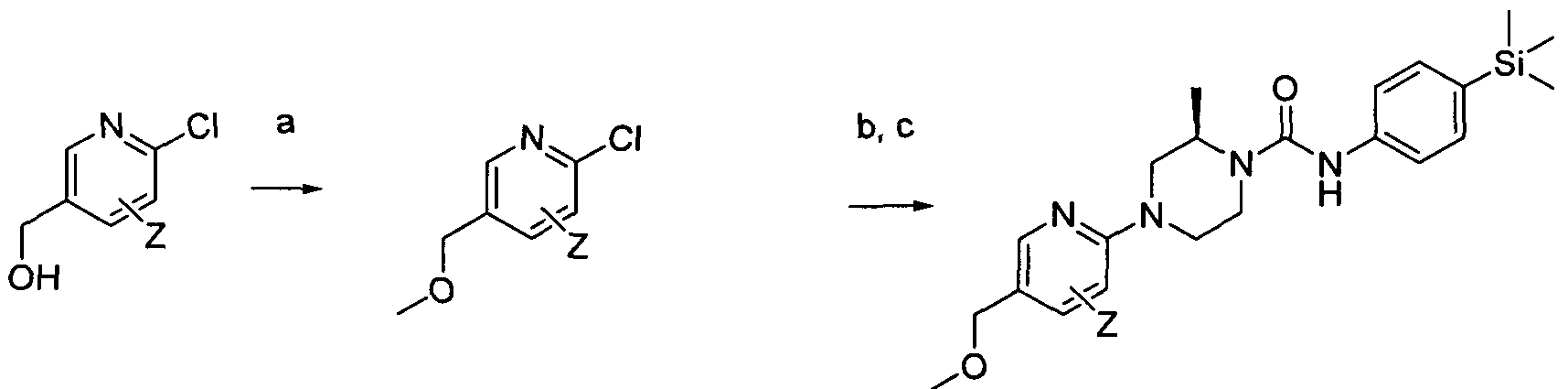
[0197]Reagents and conditions: a) THF, NaH, MeI, 16hrs b) (R) 2-methylpiperazine, TEA, DMSO, 10O0C 18 hrs c) Intermediate 1 , EtOH microwave 30 min 100 "C wh erein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0198]1.9 Scheme 9
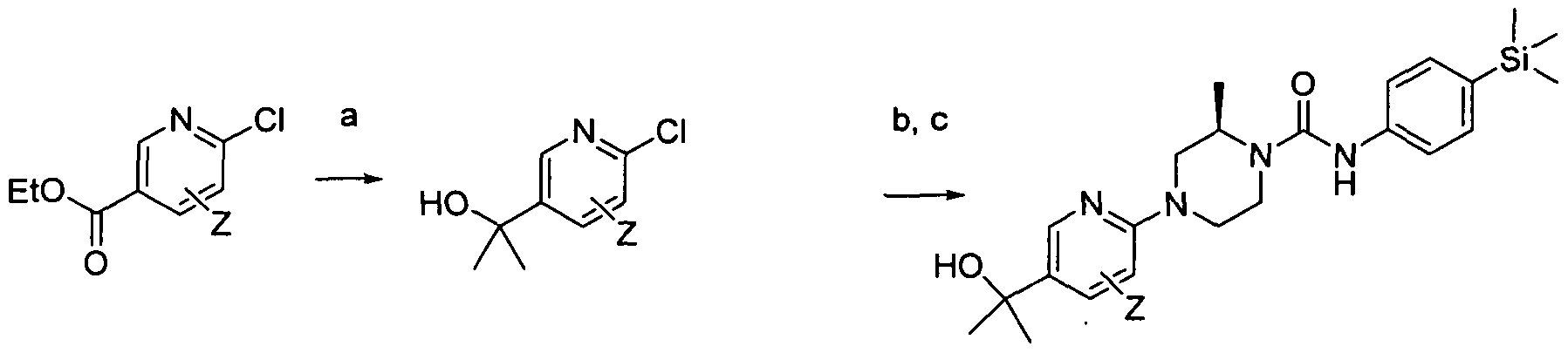
[0199]Reagents and conditions: a) THF1 -78°C, MeMgBr, 2hrs b) (R) 2-methylpiperazine, TEA, DMSO, 1000C 18 hrs c) Intermediate 1 , EtOH microwave 30 min 100 °C
[0200]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0202]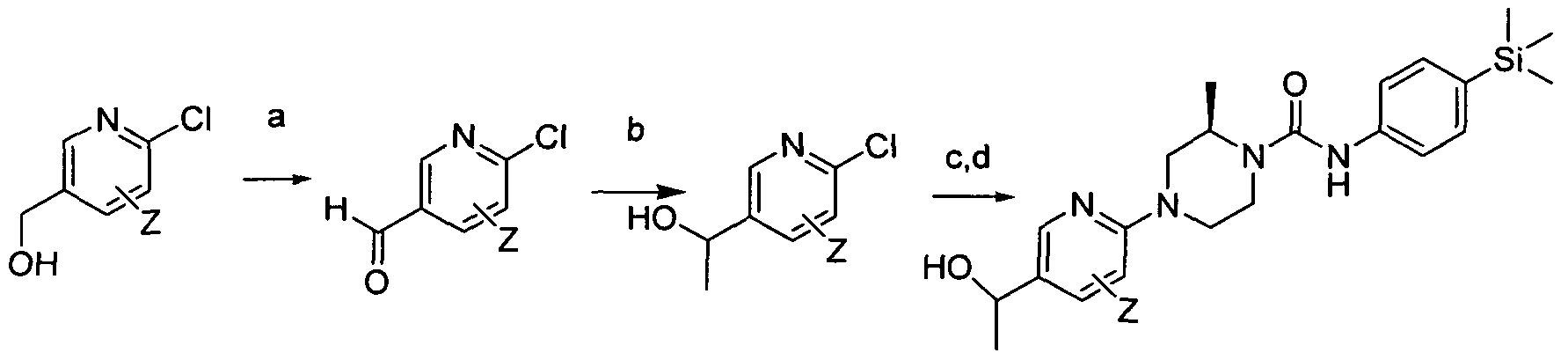
[0203]Reagents and conditions: a) MnO2, DCM, rt, 4 days b) MeMgBr, THF, -78°C c) (R) 2-methylpiperazine, TEA, DMSO, 1000C 18 hrs d) Intermediate 1 , EtOH microwave 30 min 100 °C
[0204]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0206]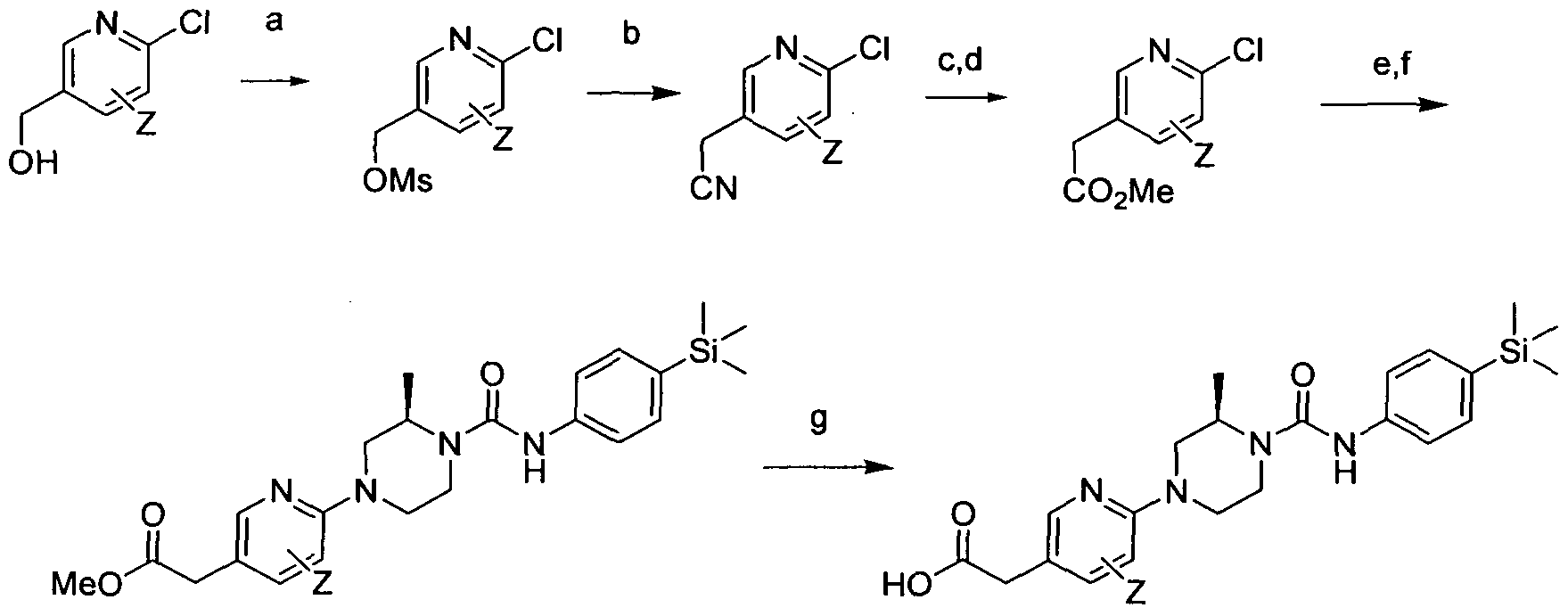
[0207]Reagents and conditions: a) TEA, DCM, MsCI, rt, 2hrs b) TEA, THF(aq), NaCN rt, 2hrs c) NaOH, 2M, microwave 100°C, 20min, d) MeOH, H2SO4(conc), rt, 2hrs, e) (R) 2-methylpiperazine, TEA, DMSO, 100°C, 18 hrs f) Intermediate 1 , EtOH microwave 30 min 1000C, g) KOH, THF(aq)
wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0209]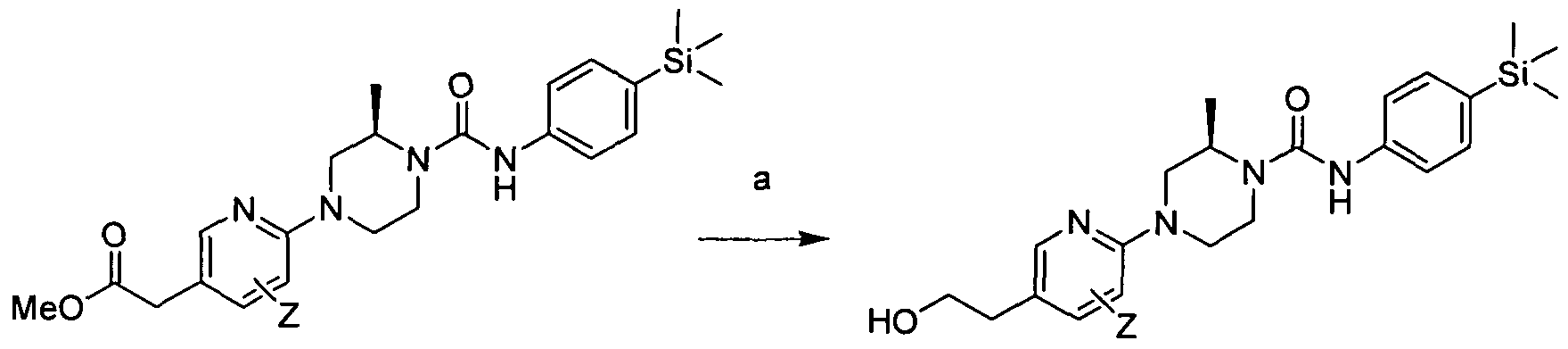
[0210]Reagents and conditions: a) THF, LiAIH4, 00C, 1hr
[0211]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio-
[0213]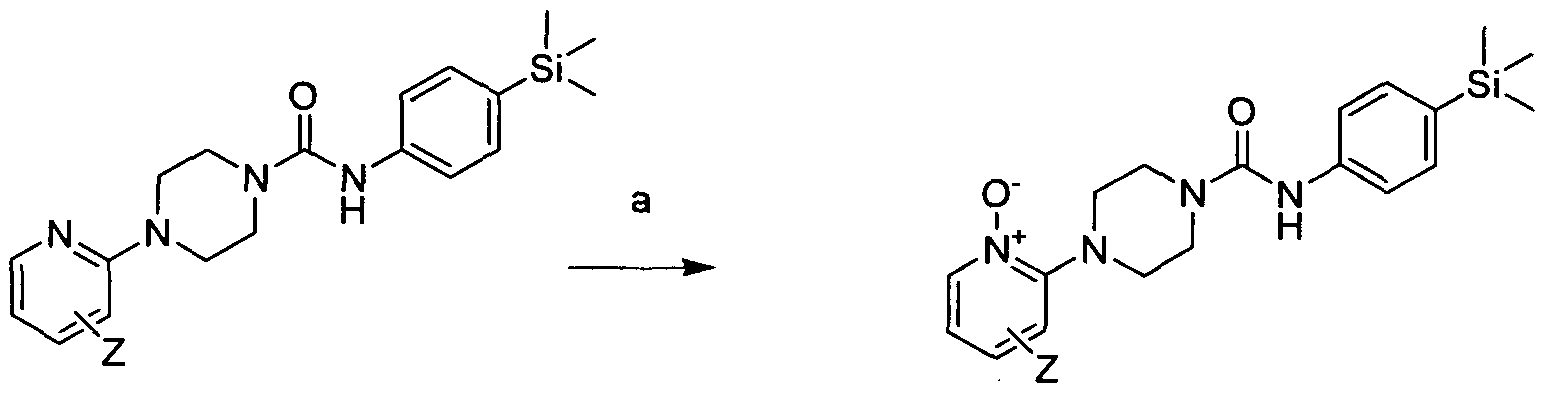
[0214]Reagents and conditions: a) NaHCO3, DCM, mCPBA, 0-50C, 1hr then 24hrs at rt
[0215]wh erein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0216]1.14 Scheme 14
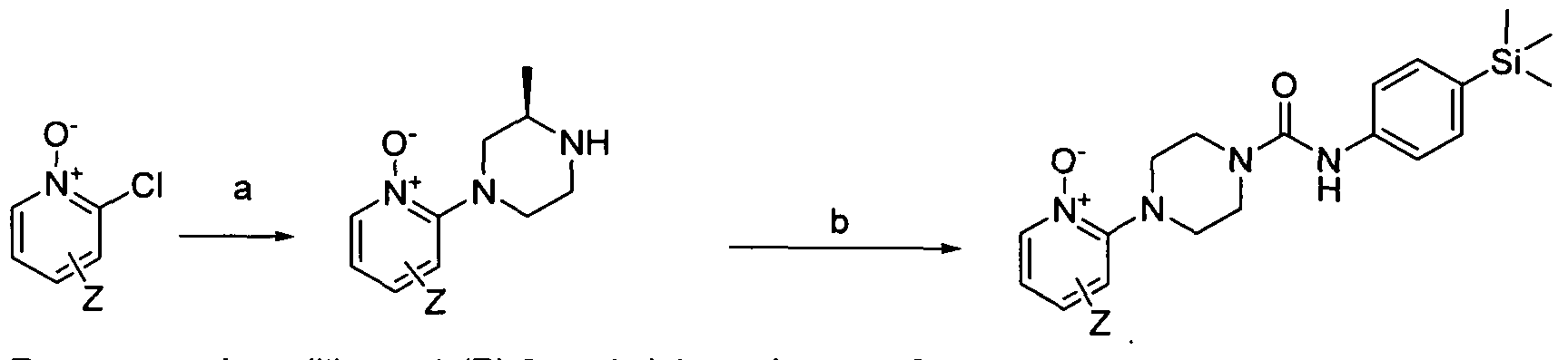
[0217]Reagents and conditions: a) (R)-2-methylpiperazine, MP-Carbonate, propan-2-ol, microwave, 150°C,90min b) Intermediate 1 , TEA, DMAP, EtOH, 100°C, microwave, 30 min whe rein Z is a group that is described by, or which can be modified to, the optional substituents described for R10 according to the first aspect of the invention.
[0219]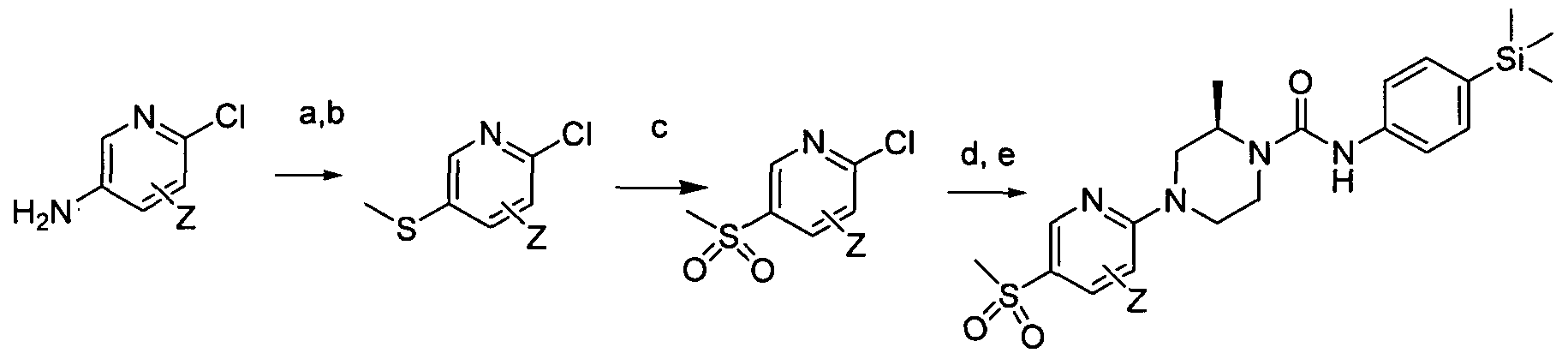
[0220]Reagents and conditions: a) NaNO2, HCI, 00C, water, 1 hr b) NaSMe, Cu(BF4J2, acetonitrile, 0°C c) mCPBA, DCM, 0°C to rt 48hr, d) (R) 2-methylpiperazine, TEA, DMSO, 1000C 18 hrs e) Intermediate 1 , EtOH microwave 30 min 100 °C
[0221]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0223]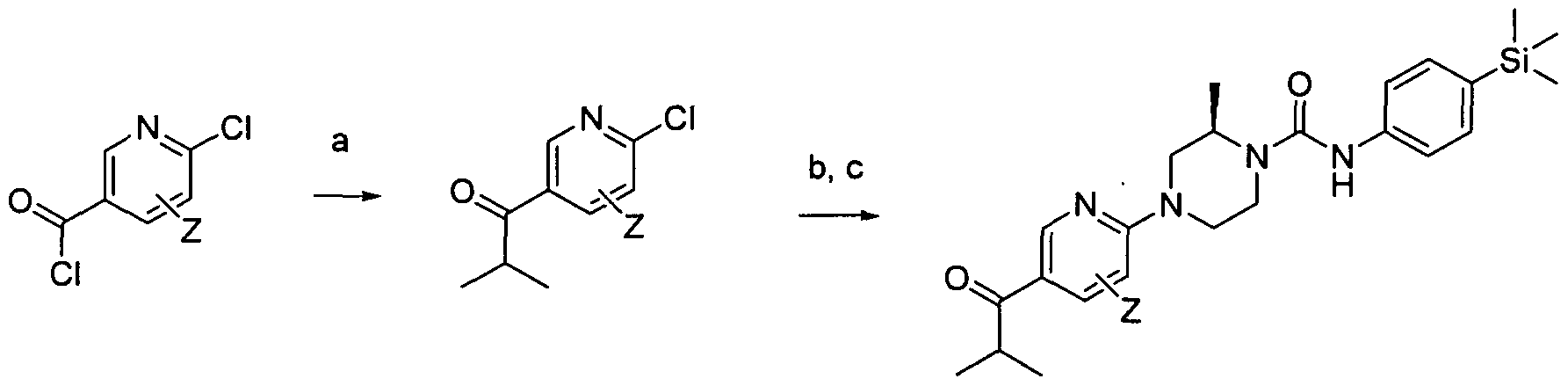
[0224]Reagents and conditions: a) 'PrMgCI, THF, -78°C b) (R) 2-methylpiperazine, TEA, DMSO, 1000C 18 hrs c) Intermediate 1 , EtOH microwave 30 min 100 °C
[0225]wherein Z is a group that is described by, or which can be modified to, the optional substituents described for Rio according to the first aspect of the invention.
[0226]2. Example Compounds
[0227]2.1 Example 1 (Prepared according to Scheme 1)
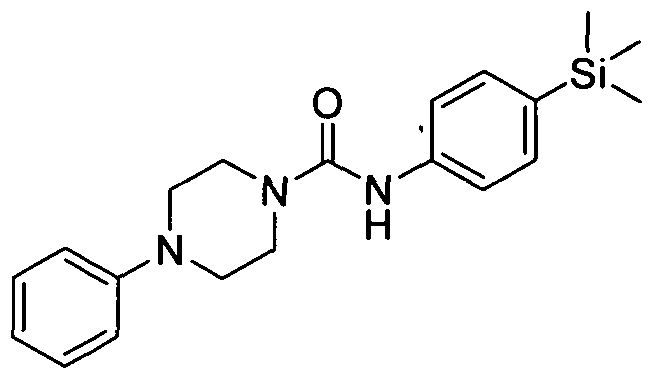
[0228]4-Phenyl-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0229]1-Phenylpiperazine (0.18mmol), Intermediate 1 (O.lδmmol) and DBU (0.53mmol) in THF (3 ml) was stirred at room temperature for 3 hrs. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (20 ml) and sodium bicarbonate (sat. 20 ml). The organic layer was separated, dried over MgSθ4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (0-100 % ethyl acetate in petroleum ether) yielding the title compound (0.029mmol). MS: ES- 352.3.1H NMR (400 MHz, DMSOd6) δ 8.44 (s, IH), 7.203 - 7.30 (m, 2H), 7.11 - 7.20 (m, 2H), 6.96 - 7.08 (m, 2H), 6.72 - 6.83 (m, 2H), 6.56 - 6.65 (m, IH), 3.35 - 3.41 (m, 4H), 2.92 - 2.99 (m, 4H), 0.00 (s, 9H)
[0230]The compounds according to Examples 2 to 12 were prepared in a similar manner to the methodology described for Example 1 and according to Scheme 1 using commercially available substituted piperazines:
[0231]2.2 Example 2 (Prepared according to Scheme 1)
[0232]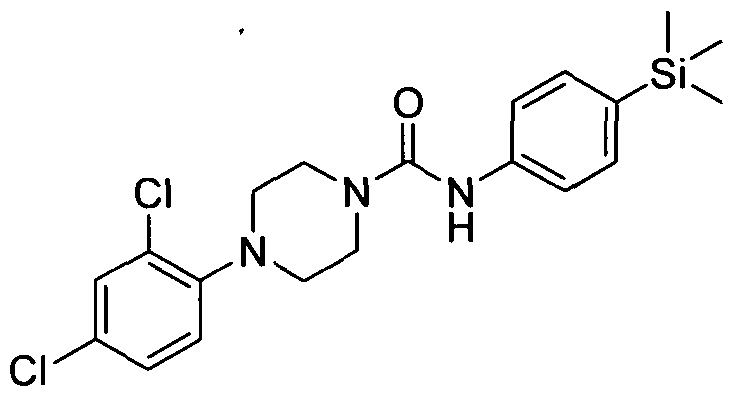
[0233]4-(2,4-Dichlorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0234]The title compound was synthesised using a procedure similar to that described for Example 1. MS: ES+ 422.0.1H NMR (400 MHz, DMSOd6) δ 7.39-7.38 (m, 2H), 7.31-7.7.30 (m, 2H) 2H), 6.90-6.88 (m, IH), 6.33 (s, IH), 3.62-3.60 (m, 4H), 3.00-2.97 (m, 4H), 0.17 (s, 9H)
[0235]2.3 Example 3 (Prepared according to Scheme 1)
[0236]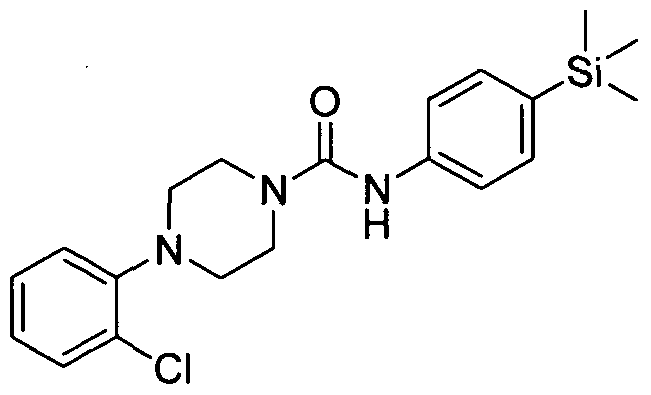
[0237]4-(2-Chlorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
The title compound was synthesised using a procedure similar to that described for Example 1. MS:ES+ 388.2.1H NMR: (400MHz, CDCl3) δ 7.45-7.43 (m, 2H), 7.38-7.36 (m, 3H), 7.28-' 7.10-7.06 (m, IH), 6.61 (s, IH) 3.67-3.66 (m, 4H), 3.07-3.06 (m, 4H), 0.24 (s, 9H)
[0238]2.4 Example 4 (Prepared according to Scheme 1)
[0239]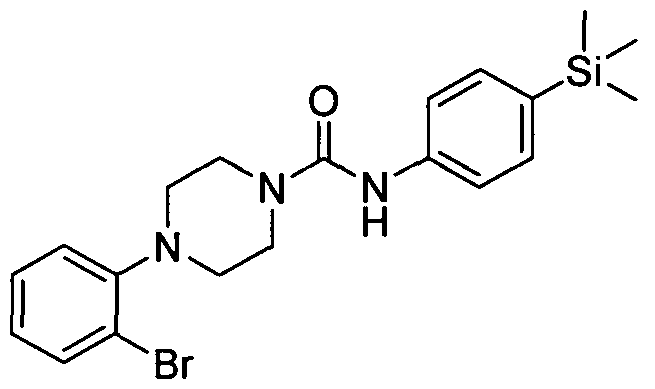
[0240]4-(2-Bromophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0241]The title compound was synthesised using a procedure similar to that described for Example 1.
[0242]MS:ES- 431.9.1H NMR (400 MHz, DMSOd6) δ 8.40 (s, IH), 7.37 - 7.43 (m, IH), 7.22 - 7.31 (m, 2H), 7.11
[0243]- 7.21 (m, 3H), 6.95 - 7.02 (m, IH), 6.74 - 6.84 (m, IH), 3.26 - 3.56 (m, 4H), 2.65 - 2.92 (m, 4H), 0.00 (s,
[0245]2.5 Example 5 (Prepared according to Scheme 1)
[0246]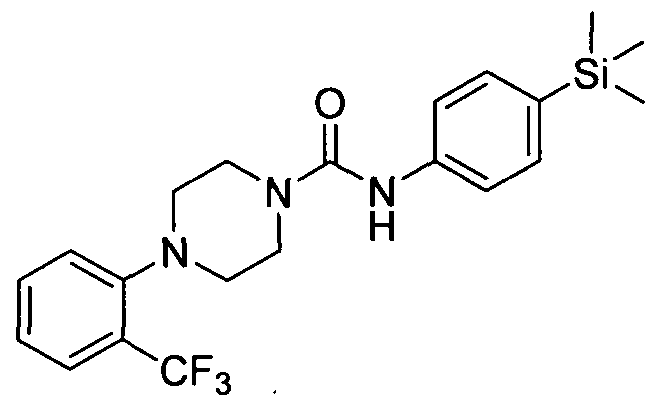 4-(2-(Trifluoromethyl)phenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
4-(2-(Trifluoromethyl)phenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0247]The title compound was synthesised using a procedure similar to that described for Example 1. MS:ES+ 421.85.1H NMR (400 MHz, DMSOd6) δ 8.38 (s, IH), 7.43 - 7.53 (m, 2H), 7.33 - 7.4 7.22 - 7.31 (m, 2H), 7.06 - 7.21 (m, 3H), 3.30 - 3.44 (m, 4H), 2.61 - 2.72 (m, 4H), 0.00 (s, 9H)
[0248]2.6 Example 6 (Prepared according to Scheme 1)
[0249]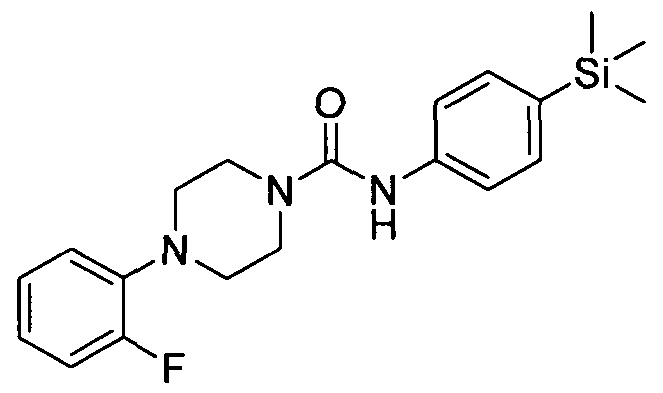
[0250]4-(2-Fluorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
The title compound was synthesised using a procedure similar to that described for Example 1. MMSS:.EESS++ 337711..99..11HH NNMMRR ((440000 M MHHzz,, DDMMSSOOdd66)) δδ 88..4411 ((ss,, IIHH)),, 77..2211 -- 77..!30 (m, 2H), 7.12 - 7.20 (m, 2H), 6.72 - 7.00 (m, 4H), 3.29 - 3.50 (m, 4H), 2.65 - 2.90 (m, 4H), 0.00 (s, 9H)
[0251]2.7 Example 7 (Prepared according to Scheme 1)
[0252]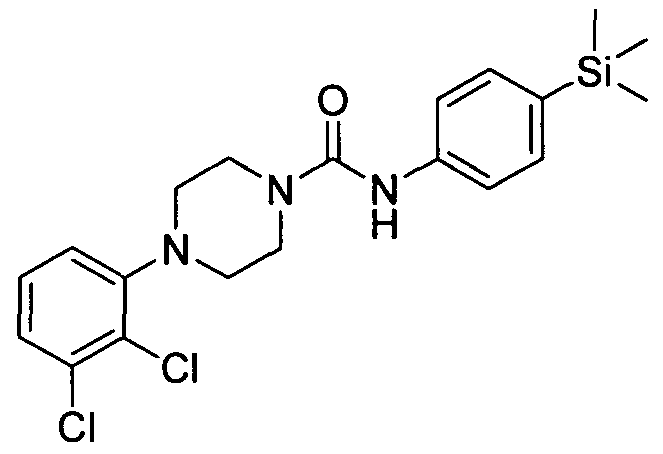
[0253]4-(2,3-Dichlorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0254]The title compound was synthesised using a procedure similar to that described for Example 1. MS:ES+ 421.9.1H NMR (400 MHz, CDCl3) δ 7.42-7.41 (m, 2H), 7.35-7.33 (m, 2H), 7.19-7.15 6.98-6.97 (m, IH), 6.44 (s, IH), 3.66-3.65 (m, 4H), 3.05-3.04 (m, 4H), 0.23 (s, 9H)
[0255]2.8 Example 8 (Prepared according to Scheme 1)
[0256]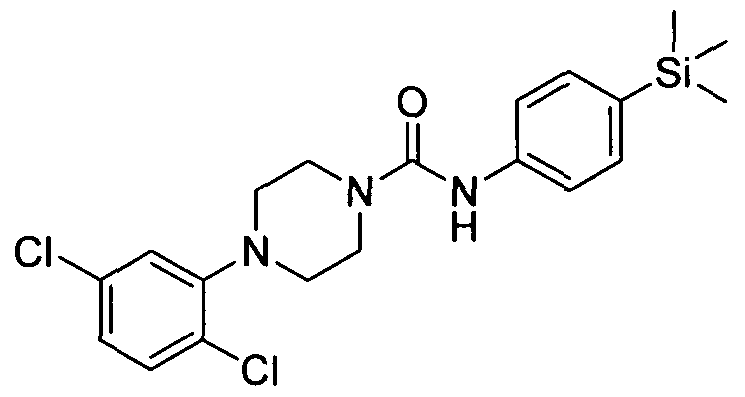 4-(2,5-Dichlorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
4-(2,5-Dichlorophenyl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0257]The title compound was synthesised using a procedure similar to that described for Example 1. MS:ES+ 422.0.1H NMR (400 MHz, CDCl3) δ 7.43-7.42 (m, 2H), 7.35-7.34 (m, 2H), 7.28-7.27 (m, IH), 6.98-6.97 (m, 2H), 6.49 (s, IH), 3.67-3.65 (m, 4H), 3.35-3.04 (m, 4H), 0.23 (s, 9H)
[0258]2.9 Example 9 (Prepared according to Scheme 1)
[0259]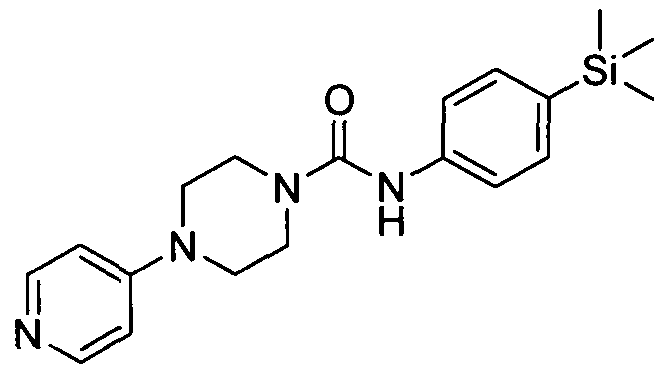
[0260]4-(Pyridin-4-yl)-N-(4-(trimethyIsilyI)phenyI)piperazine-l-carboxamide
The title compound was synthesised using a procedure similar to that described for Example 1. MS: ES+ 355.2.1H NMR (400 MHz, CDCl3) δ 8.30-8.28 (m, 2H), 7.43-7.42(m, 2H), 7.34-7.33 (m,2H), 6.64-6.63 (m, 2H), 6.57 (s, IH), 3.67-3.66 (m, 4H), 3.41-3.40 (m, 4H), 0.21 (s, 9H)
[0261]2.10 Example 10 (Prepared according to Scheme 1)
[0262]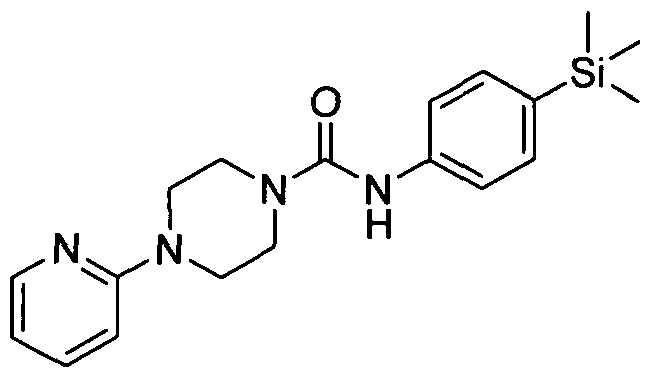
[0263]4-(Pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0264]The title compound was synthesised using a procedure similar to that described for Example 1.
[0265]MS: ES+ 354.9.1H NMR (400 MHz, DMSOd6) δ 8.41 (s, IH), 7.85 - 7.96 (m, IH), 7.30 - 7.38 (m, IH),
[0266]7.22 - 7.30 (m, 2H), 7.08 - 7.20 (m, 2H), 6.62 - 6.71 (m, IH), 6.40 - 6.50 (m, IH), 3.23 - 3.41 (m, 8H), 0.00 (s,
[0268]2.11 Example 11 (Prepared according to Scheme 1)
[0269]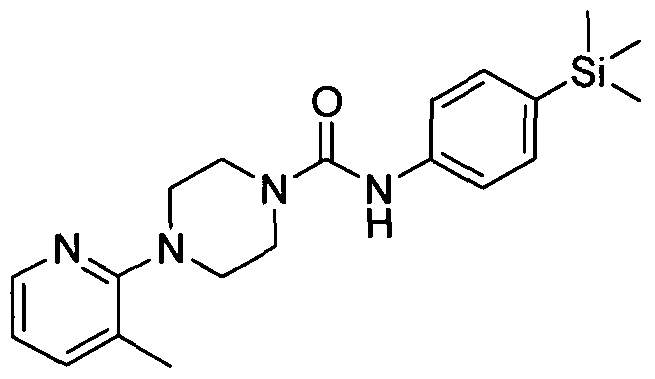
[0270]4-(3-Methylpyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0271]The title compound was synthesised using a procedure similar to that described for Example 1.
[0272]MS: ES- 367.1.1H NMR (400 MHz, DMSOd6) δ 8.39 (br. s., IH), 7.90 (br. s., IH), 7.21 - 7.42 (m, 3H),
[0273]7.01 - 7.21 (m, 2H), 6.73 (br. s., IH), 3.37 (br. s., 4H), 2.86 (br. s., 4H), 2.06 (br. s., 3H), 0.00 (br. s., 9H)
[0274]2.12 Example 12 (Prepared according to Scheme 1)
[0275]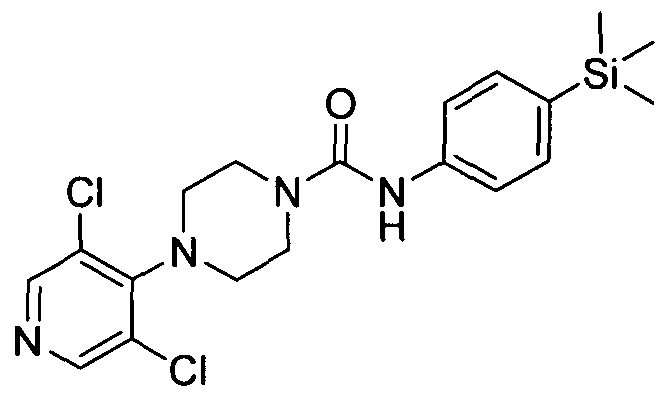
[0276]4-(3,5-Dichloropyridin-4-yl)-N-(4-(trimethyIsilyl)phenyl)piperazine-l-carboxamide
The title compound was synthesised using a procedure similar to that described for Example 1.
[0277]MS: ES+ 422.8 .1H NMR (400 MHz, DMSOd6) δ 8.43 (br. s., IH), 8.26 (s, 2H), 7.21 - 7.39 (m, 2H), 7.12
[0278]7.20 (m, 2H), 3.38 (br. s., 4H), 2.30 - 2.39 (m, 4H), 0.00 (s, 9H)
[0279]2.13 Example 13 (Prepared according to Scheme 2)
[0280]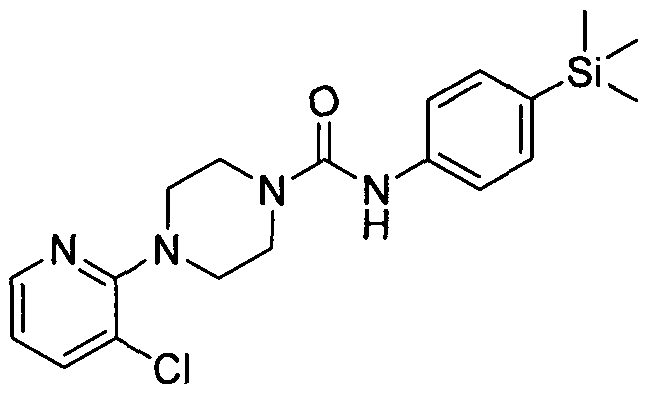
[0281]4-(3-Chloropyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0282]A mixture of piperazine (25 mmol) and 2,3-dichloropyridine (5mmol) was heated in the microwave at 1 10 °C for 30 min. The resulting residue was purified by flash chromatography (0-5 % methanol in DCM on KPNH cartridge) yielding l-(3-chloropyridin-2-yl)piperazine (3.25mmol).
[0283]1H NMR (400 MHz, CD3OD-d4) δ 7.95 (br. s., IH), 7.41 - 7.65 (m, IH), 6.59 - 6.86 (m, IH), 4.70 (br. s.,
[0284]IH), 2.87 - 3.21 (m, 4H), 2.72 - 2.89 (m, 4H) l-(3-chloropyridin-2-yl)piperazine (1.58mmol), Intermediate 1 (1.58mmol) in THF (10 ml) was heated to reflux for 18 hr. The reaction was concentrated under reduced pressure and the resulting residue was purified by flash chromatography (0-100 % ethyl acetate in petroleum ether) yielding the title compound (0.74mmol).
[0285]MS ES+ 388.8.1H NMR (400 MHz, DMSOd6) δ 8.40 (s, IH), 7.96 - 8.09 (m, IH), 7.57 - 7.72 (m, IH),
[0286]7.21 - 7.33 (m, 2H), 7.11 - 7.21 (m, 2H), 6.75 - 6.91 (m, IH), 3.28 - 3.47 (m, 4H), 3.01 - 3.09 (m, 4H), -0.13 -
[0288]2.14 Example 14 (Prepared according to Scheme 2)
[0289]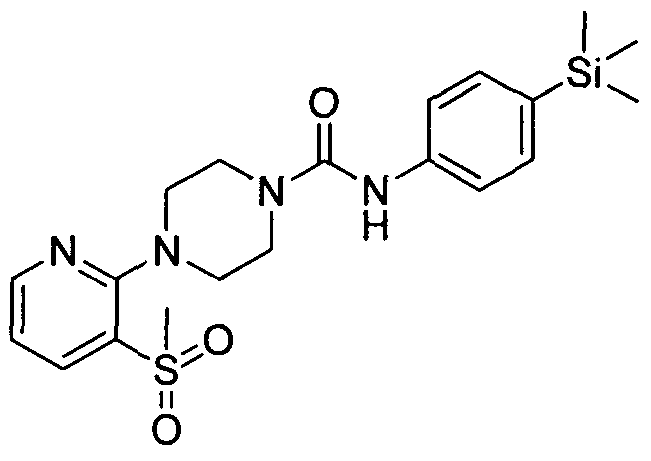 4-(3 -(Methylsulfonyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 -carboxamide
4-(3 -(Methylsulfonyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 -carboxamide
[0290]Synthesised in accordance with Example 13 from commercially available materials.
[0291]MS ES+ 433.1H NMR (400 MHz, DMSOd6) δ 8.37 - 8.48 (m, 2H), 8.07 - 8.14 (m, IH), 7.20 - 7.31 (m,
[0292]3H), 7.13 - 7.19 (m, 2H), 3.43 (br. s., 4H), 3.20 (s, 3H), 2.89 - 3.03 (m, 4H), 0.00 (s, 9H)
2.15 Example 15 (Prepared according to Scheme 1)
[0293]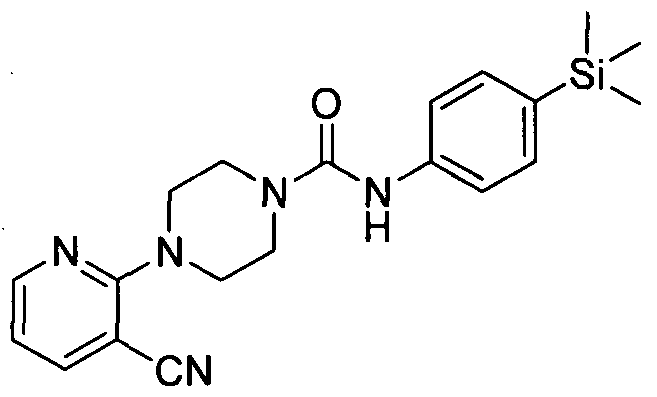
[0294]4-(3-Cyanopyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0295]Synthesised in accordance with Example 1 from commercially available materials.
[0296]MS: ES+ 379.87.1H NMR (400 MHz, DMSOd6) δ 8.39 (s, IH), 8.16 - 8.27 (m, IH), 7.84 - 7.<
[0297]7.23 - 7.31 (m, 2H), 7.07 - 7.23 (m, 2H), 6.66 - 6.82 (m, IH), 3.36 - 3.55 (m, 8H), 0.00 (s, 9H)
[0298]2.16 Example 16 (Prepared according to Scheme 1)
[0299]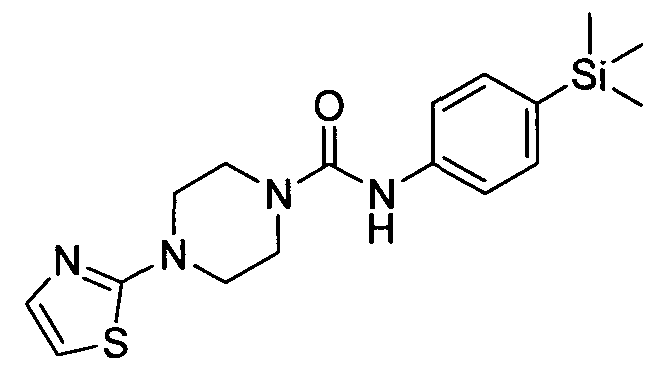 4-(Thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
4-(Thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0300]Synthesised in accordance with Example 1 from commercially available materials. MS ES+ 361.1H NMR (400 MHz, DMSOd6) δ 8.47 (s, 1 H), 7.21 - 7.30 (m, 2H), 7.12 - 7.02 (m, IH), 6.65 - 6.71 (m, IH), 3.32 - 3.44 (m, 4H), 3.19 - 3.27 (m, 4H), 0.00 (s, 9H)
[0301]2.17 Example 17 (Prepared according to Scheme 3)
[0302]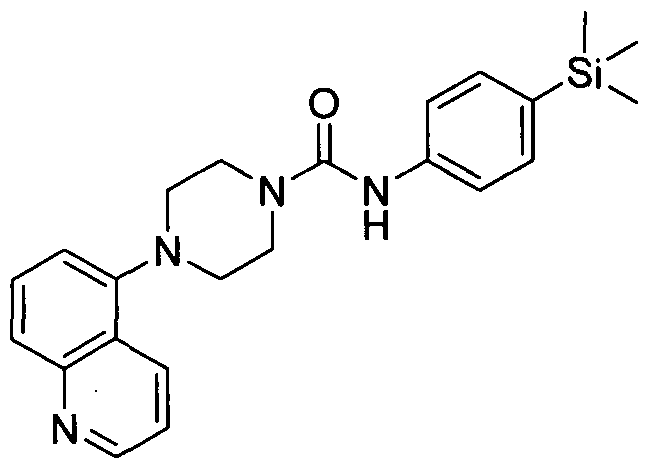
[0303]4-(Quinolin-5-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0304]5-Bromoisoquinoline (2.4 mmol), 1-Boc-piperazine (2.64 mmol), Pd(OAc)2 (0.12 mmol), BINAP (0.12 mmol) and NaO1Bu (3.36 mmol) in toluene (4 ml) was heated to 1200C in the microwave for 30 min. The
reaction mixture was poured into brine (30 ml) and extracted with ethyl acetate (30 ml). The organic phase was collected, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (20-50 % ethyl acetate in petroleum ether) yielding ter/-butyl-4-(quinolin- 5-yl)piperazine-l-caboxylate (2.11 mmol).
[0305]MS: ES+ 314.20.1H NMR (400 MHz, DMSO-d6) δ 8.89 (br. s., IH), 8.42 - 8.62 (m, IH), 7.60 - 7.84 (m, 2H), 7.42 - 7.60 (m, IH), 7.15 - 7.34 (m, IH), 3.61 (br. s., 4H), 2.99 (br. s., 4H), 1.44 (s, 9H)
[0306]To a solution of fer/-butyl-4-(quinolin-5-yl)piperazine-l-carboxylate (0.88 mmol) in 1,4-dioxane (10 ml) and MeOH (2 ml) was added 4M HCl in dioxane (4.38 mmol). The reaction was stirred at room temperature for 18 hrs. The reaction was concentrated under reduced pressure yielding 5-(piperazin-l-yl)quinoline hydrochloride (0.88 mmol).
[0307]MS: ES+ 214.30.1H NMR (400 MHz, DMSO-d6) δ 9.85 (br. s., IH), 9.61 (br. s., 2H), 8.40 - 8.85 (m, 2H), 8.19 (br. s., IH), 7.69 - 7.98 (m, 2H), 3.20 - 3.54 (m, 8H)
[0308]A solution of 5-(piperazin-l-yl)quinoline hydrochloride (0.42mmol), Intermediate 1 (0.35mmol), and DBU (1.05mmol) in THF (5 ml) was stirred at room temperature for 2 hrs. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (20 ml) and sodium bicarbonate (sat. 20 ml). The organic phase was separated, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (50-100% ethyl acetate in petroleum ether) yielding the title compound (O.lόmmol).
[0309]MS: ES+ 405 .1H NMR (400 MHz, DMSO-d6) δ 8.63 - 8.73 (m, IH), 8.47 (s, IH), 8.30 - 8.40 (m, IH), 7.41 - 7.59 (m, 2H), 7.30 - 7.39 (m, IH), 7.23 - 7.30 (m, 2H), 7.13 - 7.21 (m, 2H), 6.98 - 7.08 (m, IH), 3.53 (br. s., 4H), 2.84 (br. s., 4H), 0.00 (s, 9H)
[0310]2.18 Example 18 (Prepared according to Scheme 3)
[0311]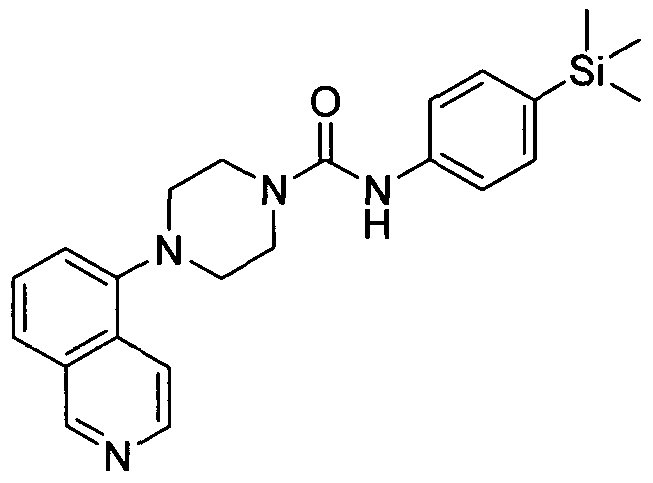
[0312]4-(Isoquinolin-5-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0313]Synthesised in accordance with Example 17
[0314]MS ES+ 405.1H NMR (400 MHz, DMSO-d6) δ 9.08 (s, IH), 8.48 (s, IH), 8.29 - 8.34 (m, IH), 7.74 - 7.80 (m, IH), 7.54 - 7.64 (m, IH), 7.35 - 7.45 (m, IH), 7.23 - 7.32 (m, 2H), 7.13 - 7.21 (m, 3H), 3.53 (br. s., 4H), 2.85 (br. s., 4H), 0.00 (s, 9H)
2.19 Example 19 (Prepared according to Scheme 2)
[0315]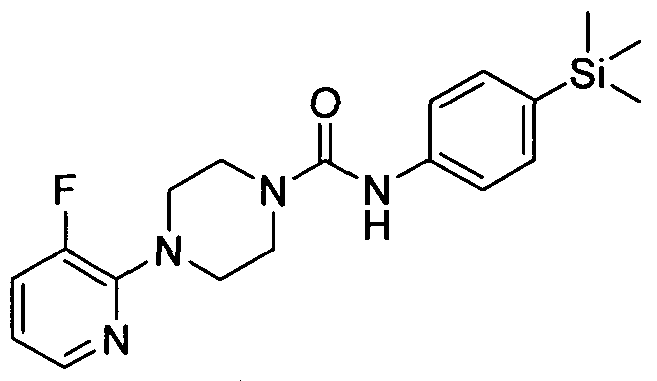
[0316]4-(3 -Fluoropyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 -carboxamide
[0317]Synthesised in accordance with Example 13 from commercially available materials
[0318]MS: ES+ 373.12.1H NMR (400 MHz, DMSOd6) δ 8.42 (s, IH), 7.73 - 7.90 (m, IH), 7.29 - 7.41 (m, IH),
[0319]7.21 - 7.29 (m, 2H), 7.08 - 7.20 (m, 2H), 6.65 - 6.81 (m, IH), 3.38 (br. s., 4H), 3.20 (br. s., 4H), 0.00 (s, 9H)
[0320]2.20 Example 20 (Prepared according to Scheme 1)
[0321]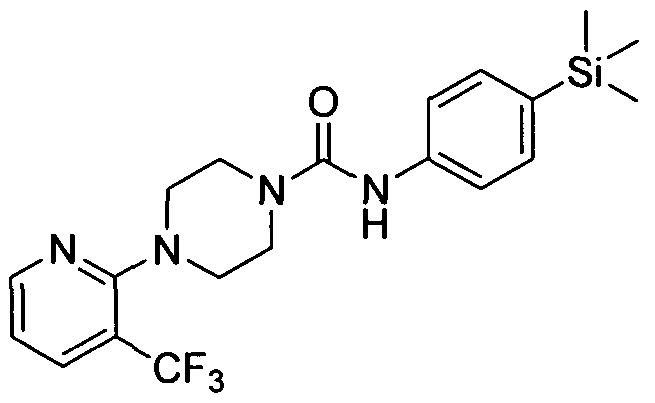 4-(3-(Trifluoromethyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
4-(3-(Trifluoromethyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0322]Synthesised in accordance with Example 1 from commercially available materials
[0323]MS:ES+ 422.85.1H NMR (400 MHz, DMSOd6) δ 8.39 (s, IH), 8.30 - 8.37 (m, IH), 7.85 - 7.96 (m, IH),
[0324]7.21 - 7.33 (m, 2H), 7.12 - 7.21 (m, 2H), 6.98 - 7.07 (m, IH), 3.33 - 3.45 (m, 4H), 2.93 - 3.04 (m, 4H), 0.00 (s,
[0326]2.21 Example 21 (Prepared according to Scheme 1)
[0327]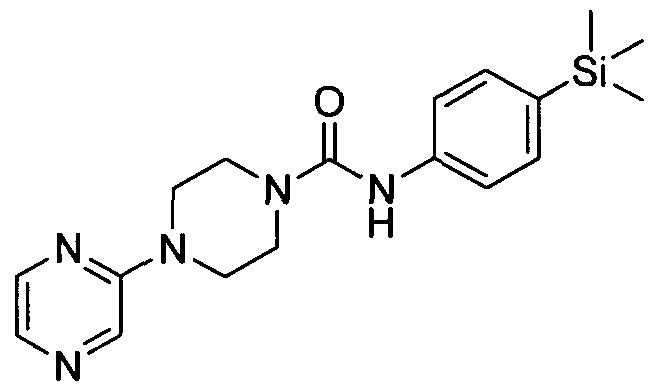
[0328]4-(Pyrazin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0329]Synthesised in accordance with Example 1 from commercially available materials. MS: ES+ 355.9.1H NMR (400 MHz, DMSOd6) δ 8.43 (s, IH), 8.15 (s, IH), 7.89 (br. s., IH), 7.60 - 7.69 (m, IH), 7.21 - 7.29 (m, 2H), 7.10 - 7.21 (m, 2H), 3.27 - 3.61 (m, 8H), 0.20 - 0.17 (m, 9H)
2.22 Example 22 (Prepared according to Scheme 1)
[0330]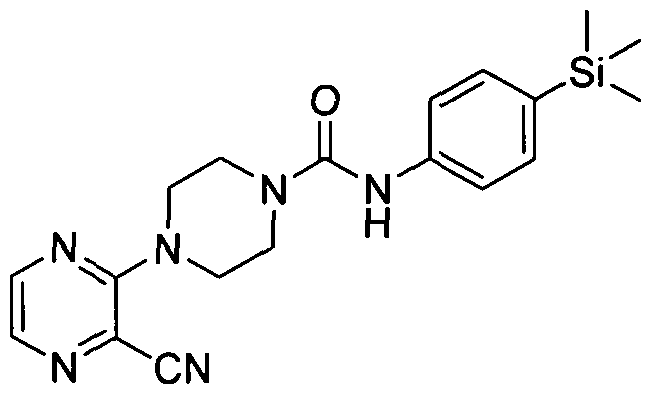
[0331]4-(3-Cyanopyrazin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0332]Synthesised in accordance with Example 1 from commercially available materials.
[0333]MS: ES+ 381.14.1H NMR (400 MHz, DMSOd6) δ 8.42 (s, IH), 8.21 - 8.31 (m, IH), 7.91 - 7.95 (m, IH)5
[0334]7.22 - 7.31 (m, 2H), 7.11 - 7.20 (m, 2H), 3.54 - 3.63 (m, 4H), 3.42 (br. s., 4H), 0.00 (s, 9H)
[0335]2.23 Example 23 (Prepared according to Scheme 4)
[0336]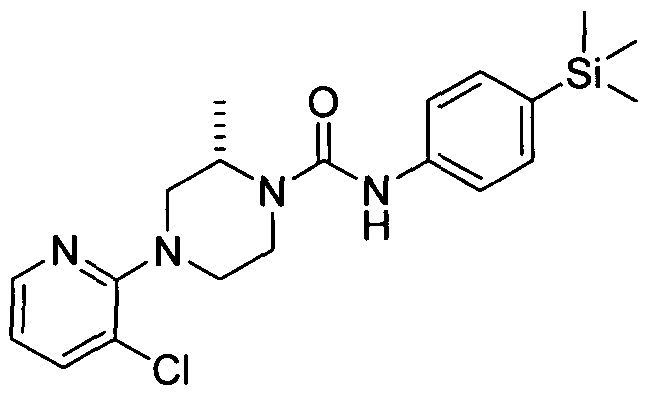
[0337](S)-4-(3-Chloropyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0338]A solution of 2,3-dichloropyridine (3mmol), (5)-(+)-2-methylpiperazine (3mmol) and TEA (9mmol) in DMSO (20 ml) was heated to 100 °C for 18 hrs. The reaction mixture was diluted with MeOH (20 ml) and put onto an SCX cartridge. The cartridge was washed with MeOH before eluting the product off with 2M ammonia in MeOH. The appropriate fractions were collected and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (60-80 % ethyl acetate in petroleum ether on KPNH cartridge) yielding (S)-l-(3-chloropyridine-2-yl)-3-methylpiperazine (2.1mmol).
[0339]MS: ES+ 212.10.1H NMR (400 MHz, DMSO-d6) δ 8.14 - 8.28 (m, IH), 7.67 - 7.87 (m, IH), 6.84 - 7.08 (m, IH), 3.49 - 3.62 (m, 2H), 2.87 - 2.96 (m, IH), 2.77 - 2.87 (m, 2H), 2.62 - 2.77 (m, IH), 2.51 (s, IH), 2.34 - 2.44 (m, IH), 0.92 - 1.10 (m, 3H)
[0340]A solution of (,S>-l-(3-chloropyridine-2-yl)-3-methylpiperazine (0.53mmol) and Intermediate 1 (0.53mmol) in ethanol (2 ml) was heated in the microwave to 1000C for 30 min. The reaction mixture was concentrated under educed pressure and the resulting residue was purified by flash chromatography (20-40 % ethyl acetate in petroleum ether) yielding the title compound (0.32 mmol).
[0341]MS ES- 401.20.1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, IH), 8.00 - 8.07 (m, IH), 7.57 - 7.66 (m, IH), 7.22 - 7.30 (m, 2H), 7.1 1 - 7.20 (m, 2H), 6.78 - 6.88 (m, IH), 4.26 (br. s., IH), 3.77 (br. s., IH), 3.41 - 3.52 (m, 2H), 2.98 - 3.08 (m, IH), 2.55 - 2.77 (m, 2H), 1.04 - 1.13 (m, 3H), 0.00 (s, 9H)
2.24 Example 24 (Prepared according to Scheme 4)
[0342]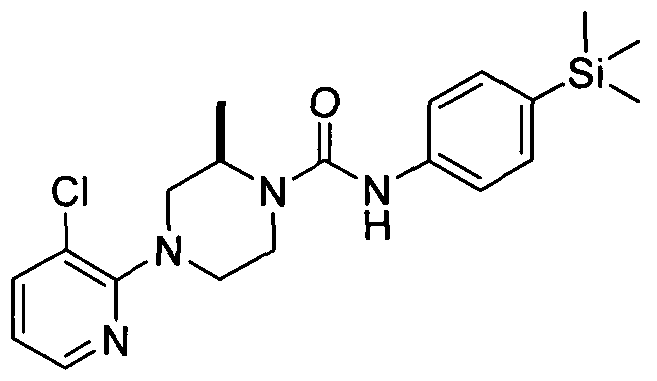
[0343](^^-(S-Dichloropyridin-Z-yO^-methyl-N^-CtrimethylsilyOphenyOpiperazine-l-carboxamide
[0344]Synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine.
[0345]MS: ES+ 403.10.1H NMR (400 MHz, DMSOd6) δ 8.34 (br. s., IH), 7.94 - 8.10 (m, IH), 7.54 - 7.70 (m, IH), 7.21 - 7.34 (m, 2H), 7.09 - 7.21 (m, 2H), 6.78 - 6.89 (m, IH), 4.17 - 4.31 (m, IH), 3.72 - 3.85 (m, IH), 3.36 - 3.54 (m, 2H), 2.97 - 3.07 (m, IH), 2.54 - 2.79 (m, 2H), 0.94 - 1.27 (m, 3H), 0.00 (br. s., 9H)
[0346]2.25 Example 25 (Prepared according to Scheme 4)
[0347]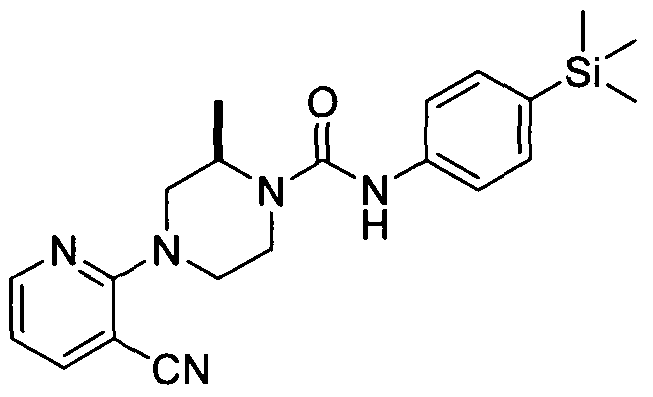 (R)-4-(3-Cyanopyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
(R)-4-(3-Cyanopyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0348]Synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine.
[0349]MS ES+ 378.8.1H ΝMR (400 MHz, DMSOd6) δ 8.99 (br. s., IH), 7.99 (d, J= 7.07 Hz, IH), 7.79 - 7.92 (m, 2H), 7.75 (d, J= 6.32 Hz, 3H), 7.58 (d, J= 6.57 Hz, IH), 7.32 - 7.45 (m, IH), 3.99 (br. s., 4H), 3.36 (br. s., 4H), 0.59 (br. s., 9H)
[0350]2.26 Example 26 (Prepared according to Scheme 4)
[0351]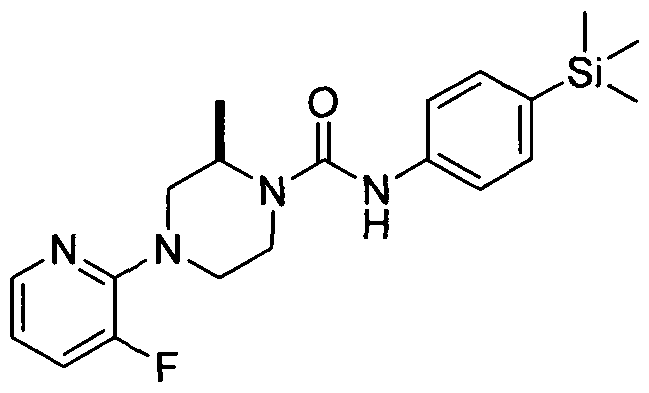 (/?)-4-(3-Fluoropyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
(/?)-4-(3-Fluoropyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0352]Synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine .
MS ES+ 387.1H NMR (400 MHz, DMSOd6) δ 8.32 (s, IH), 7.74 - 7.89 (m, IH), 7.21 - 7.41 (m, 3H), 7.07 • 7.22 (m, 2H), 6.62 - 6.77 (m, IH), 4.24 (br. s., IH), 3.69 - 3.85 (m, 2H), 3.55 - 3.64 (m, IH), 2.96 - 3.09 (m, IH), 2.81 - 2.92 (m, IH), 2.58 - 2.75 (m, IH), 0.93 - 1.09 (m, 3H), 0.00 (s, 9H)
[0353]2.27 Example 27 (Prepared according to Scheme 4)
[0354]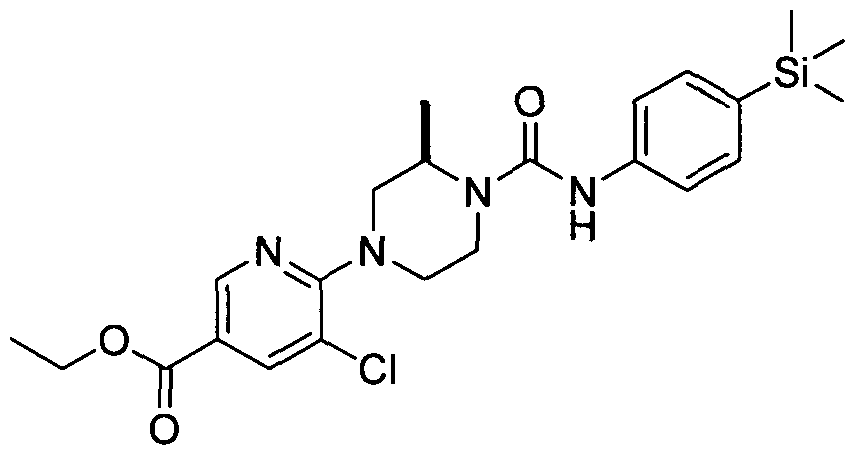
[0355](Λ)-Ethyl 5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin- 1 -yl)nicotinate
[0356]Synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine.
[0357]MS ES+ 475.1.1H NMR (400 MHz, DMSOd6) δ 8.43 - 8.51 (m, IH), 8.32 (s, IH), 7.85 - 7.94 (m, IH), 7.21 - 7.29 (m, 2H), 7.11 - 7.19 (m, 2H), 4.20 - 4.31 (m, IH), 4.03 - 4.15 (m, 2H), 3.73 - 3.87 (m, 3H), 3.09 - 3.16 (m, IH), 2.90 - 3.00 (m, IH), 2.72 - 2.85 (m, IH), 1.06 - 1.15 (m, 3H), 0.96 - 1.04 (m, 3H), 0.00 (s, 9H)
[0358]2.28 Example 28 (Prepared according to Scheme 5, Step a)
[0359]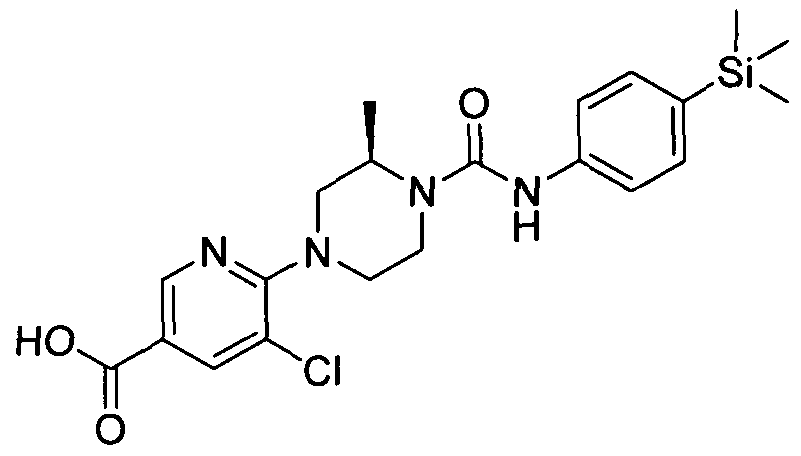 (R)-5-Chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
(R)-5-Chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
[0360]To a solution of (R)-Ethyl5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l- yl)nicotinate (Example 27) (11.2mmol) in THF (150ml) and water (50 ml) was added potassium hydroxide (33.5mmol). The reaction mixture was stirred at ambient temperature for 60 hours. The THF was removed under reduced pressure and the resulting suspension diluted with sodium bicarbonate (sat., 150 ml) and extracted with ethyl acetate (150 ml). The aqueous was separated and acidified to pH 1 with 2N hydrochloric acid and extracted with ethyl acetate (2 xlOOml). The organics were combined, dried (MgSO4) and concentrated under reduced pressure. The resulting off white solid was triturated with methanol (cold), filtered and dried yielding the title compound in 60 % yield.
[0361]MS ES+ 447.3.1H NMR (400 MHz, CDCl3) δ 8.78 - 8.86 (m, IH), 8.16 - 8.25 (m, IH), 7.42 - 7.51 (m, 2H), 7.33 - 7.40 (m, 2H), 6.47 (s, IH), 4.35 - 4.46 (m, IH), 4.06 - 4.23 (m, 2H), 3.89 - 4.01 (m, IH), 3.44 - 3.58 (m, 2H), 3.27 - 3.37 (m, IH), 3.06 - 3.20 (m, IH), 1.34 - 1.46 (m, 3H), 0.25 (s, 9H)
2.29 Example 29 (Prepared according to Scheme 5 step c)
[0362]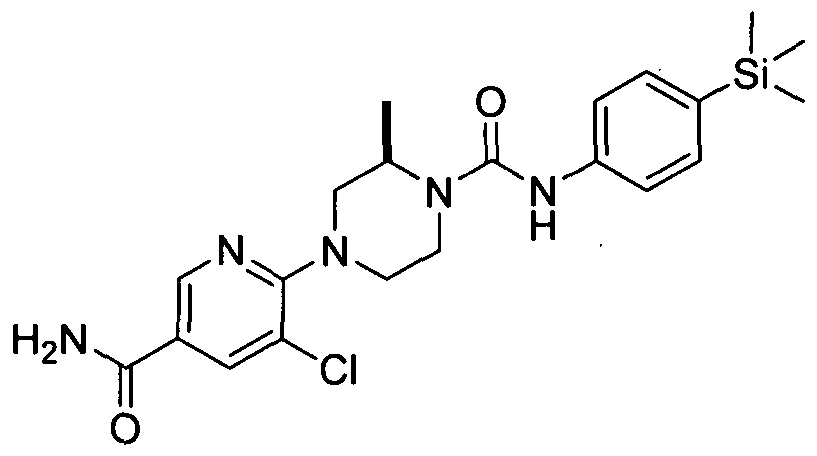 (/?)-4-(5-Carbamoyl-3-chloropyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
(/?)-4-(5-Carbamoyl-3-chloropyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0363]To a solution of (Λ)-Ethyl 5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l- yl)nicotinate (Example 27) (0.32mmol), ammonium chloride (0.32mmol) and /ert-butylammonium bromide (0.32mmol) in ammonium hydroxide (3.2ml) was stirred at room temperature for 3 days. The reaction was concentrated under reduced pressure and the resulting residue was purified by flash chromatography (0- 100 % ethyl acetate in petroleum ether) yielding the title compound (O.lOόmmol). MS:ES+ 446.0.1H NMR (400 MHz, DMSOd6) δ 8.42 - 8.48 (m, IH), 8.32 (s, IH), 7.93 - 8.01 (m, IH), 7.79 (br. s., IH), 7.21 - 7.30 (m, 3H), 7.12 - 7.19 (m, 2H), 4.21 - 4.32 (m, IH), 3.74 - 3.84 (m, IH), 3.57 - 3.73 (m, 2H), 3.00 - 3.07 (m, IH), 2.80 - 2.91 (m, IH), 2.64 - 2.77 (m, IH), 1.01 - 1.07 (m, 3H), 0.00 (s, 9H)
[0364]2.30 Example 30 (Prepared according to Scheme 5 step b)
[0365]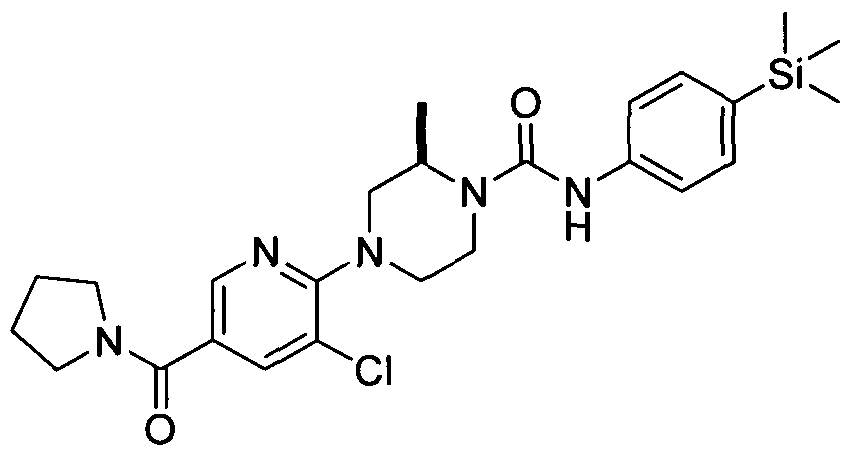
[0366](R)-4-(3-Chloro-5-(pyrrolidine-l-carbonyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0367]To a solution of (R)-5-Chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l -yl)nicotinic acid (Example 28) (0.34mmol), pyrrolidine (0.37mmol), EDC (0.50mmol), and TEA (1.Olmmol) in DCM (3 ml) was stirred at room temperature for 18 hrs. The reaction was concentrated under reduced pressure and the resulting residue was purified by flash chromatography (40-100 % ethyl acetate in petroleum ether) yielding the title compound (O.lOmmol).
[0368]MS:ES+ 500.1.1H NMR (400 MHz, DMSOd6) δ 8.32 (s, IH), 8.07 - 8.24 (m, IH), 7.68 - 7.82 (m, IH), 7.21 - 7.32 (m, 2H), 7.06 - 7.21 (m, 2H), 4.14 - 4.35 (m, IH), 3.71 - 3.89 (m, IH), 3.54 - 3.69 (m, 2H), 3.18 - 3.37 (m, 4H), 2.58 - 2.87 (m, 2H), 1.63 (br. s., 4H), 1.38 - 1.51 (m, IH), 0.98 - 1.13 (m, 3H), 0.00 (s, 9H)
2.31 Example 31 (Prepared according to Scheme 6, step a)
[0369]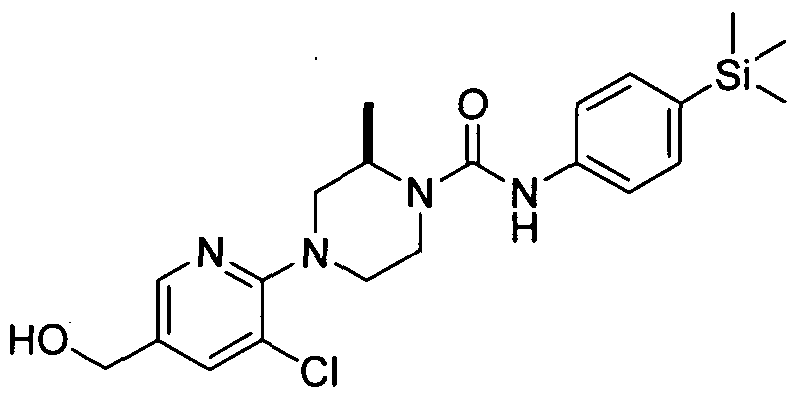
[0370](R)-4-(3-Chloro-5-(hydroxymethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl) piperazine-l- carboxamide
[0371]To a solution of (R)-Ethyl 5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l- yl)nicotinate (Example 27) (2.11mmol) in EtOH (50 ml) was added lithium borohydride (2.32mmol) portionwise. The reaction was stirred at room temperature for 48 hrs. The reaction was filtered through a pad of celite® and concentrated under reduced pressure. The resulting waxy solid was purified by flash chromatography (50-100 % ethyl acetate in petroleum ether) yielding the title compound (1.47mmol). MS:ES- 431.2.1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, IH), 7.96 (s, IH), 7.43 - 7.59 (m, IH), 7.20 - 7.32 (m, 2H), 7.06 - 7.10 (m, OH), 7.04 - 7.20 (m, 2H), 4.93 - 5.15 (m, IH), 4.16 - 4.29 (m, 3H), 3.65 - 3.92 (m, IH), 3.33 - 3.48 (m, 2H), 3.02 (br. s., IH), 2.54 - 2.75 (m, 2H), 1.05 - 1.13 (m, 3H), 0.00 (s, 9H)
[0372]2.32 Example 32 (Prepared according to Scheme 4)
[0373]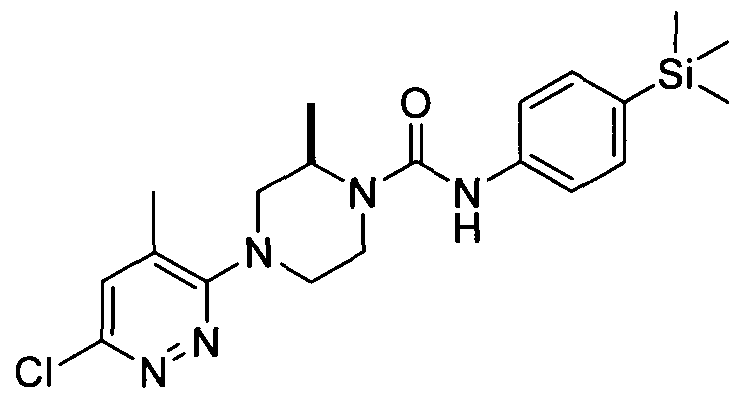
[0374](R)-4-(6-Chloro-4-methylpyridazin-3-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0375]A solution of 3,6-dichloro-4-methylpyridazine (6.13mmol), (R)-2-methylpiperazine (6.13mmol) and TEA (18.4mmol) in DMSO (30 ml) was heated to 1000C for 18hrs. The reaction was diluted with MeOH (20 ml) and put onto an SCX cartridge. The cartridge was washed with methanol and the product was eluted with 2M ammonia in methanol. The appropriate fractions were concentrated under reduced pressure. The resulting residue was purified by flash chromatography (50-100 % ethyl acetate in petroleum ether on KPNH cartridge) yielding (Λ)-6-chloro-4-methyl-3-(3-methylpiperazin-l-yl)pyridazine (1.21mmol) and (R)-3- chloro-4-methyl-6-(3-methylpiperazin-l-yl)pyridazine (3.63mmol) inseparable by chromatography. 1H NMR (400 MHz, DMSO-d6) δ 7.22 - 7.67 (m, IH), 4.08 - 4.19 (m, IH), 3.27 - 3.47 (m, IH), 2.59 - 3.06 (m, 4H), 2.34 - 2.48 (m, IH), 2.27 (s, IH), 0.92 - 1.12 (m, 3H)
[0376]A solution of (Λ)-6-chloro-4-methyl-3-(3-methylpiperazin-l-yl)pyridazine and (Λ)-3-chloro-4-methyI-6-(3- methylpiperazin-l-yl)pyridazine (4.86mmol) and Intermediate 1 (4.86mmol) in EtOH (20 ml) was heated to
1000C in the microwave for 30 min. The reaction was concentrated under reduced pressure. The residue was purified by flash chromatography (20-50 % ethyl acetate in petroleum ether) yielding the title compound
[0378]MS: ES+ 418.1.1H NMR (400 MHz, DMSOd6) δ 8.35 (s, IH), 7.43 (s, IH), 7.23 - 7.28 (m, 2H), 7.11 - 7.19
[0379](m, 2H), 4.20 - 4.33 (m, IH), 3.75 - 3.85 (m, IH), 3.33 - 3.42 (m, IH), 3.20 - 3.30 (m, IH), 3.03 - 3.06 (m,
[0380]IH), 2.61 - 2.85 (m, 2H), 2.14 (s, 3H), 1.04 - 1.12 (m, 3H), 0.00 (s, 9H)
[0381]2.33 Example 33 (Prepared according to Scheme 6, steps b and c)
[0382]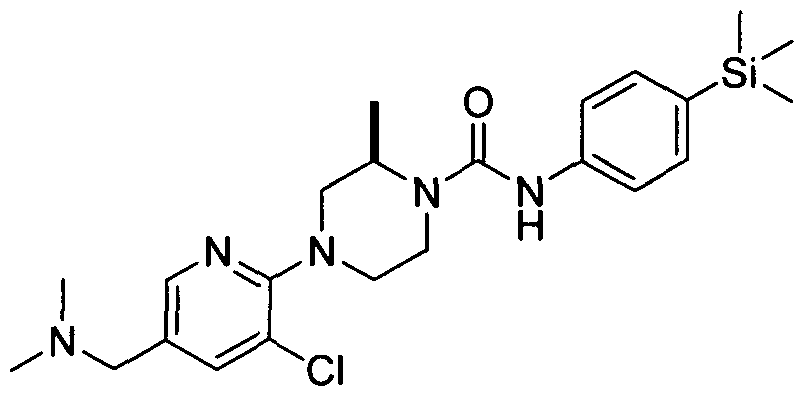
[0383](Λ)-4-(3-Chloro-5-((dimethylamino)methyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0384]To a solution of (/?)-4-(3-Chloro-5-(hydroxymethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl) piperazine-1 -carboxamide (Example 31) (0.46mmol), and TEA (0.92mmol) in DCM (10 ml) was added MsCl (0.46mmol) in a dropwise fashion. The reaction was stirred at room temperature for 1 hr. The reaction was diluted with DCM (10 ml) and washed with sodium bicarbonate (sat, 20 ml). The organic layer was separated, dried over MgSO4, filtered and concentrated under reduced pressure yielding (i?)-(5-chloro-6-(3- methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridine-3-yl)methyl methanesulfonate
[0386]1H NMR (400 MHz, CDCl3) δ 8.17 - 8.21 (m, IH), 7.67 - 7.72 (m, IH), 7.44 - 7.49 (m, 2H), 7.38 - 7.44 (m, 2H), 4.54 (s, 2H), 4.36 - 4.45 (m, IH), 3.79 - 4.01 (m, 3H), 3.69 (s, IH), 3.40 - 3.53 (m, IH), 3.20 - 3.30 (m, IH), 3.14 - 3.19 (m, 2H), 3.08 - 3.14 (m, IH), 2.97 - 3.06 (m, IH), 1.42 - 1.47 (m, 3H), 0.23 - 0.33 (m, 9H)
[0387]A solution of (RHS-chloro-ό^-methyl^^-^rimethylsily^phenylcarbamoyOpiperazin-l-yOpyridine^- yl)methyl methanesulfonate (0.26mmol), dimethylamine (2M in THF, 0.52mmol) and TEA (0.39mmol) in THF (3 ml) was stirred at room temperature for 2 hrs. The reactions were concentrated under reduced pressure and the residue was purified by flash chromatography (0-100 % ethyl acetate in petroleum ether) yielding the title compound (0.089mmol).
[0388]MS: ES+ 460.1.1H NMR (400 MHz, CDCl3) δ 7.92 - 8.02 (m, IH), 7.52 - 7.59 (m, IH), 7.32 - 7.39 (m, 2H), 7.23 - 7.31 (m, 2H), 6.25 (s, IH), 4.22 - 4.35 (m, IH), 3.82 (d, J= 12.63 Hz, OH), 3.62 - 3.76 (m, 2H), 3.31 - 3.44 (m, OH), 3.25 (s, 2H), 2.83 - 3.04 (m, 2H), 2.14 (s, 6H), 1.46 (s, 2H), 1.30 - 1.41 (m, 3H), 0.15 (s, 9H)
[0389]2.34 Example 34 (Prepared according to Scheme 4)
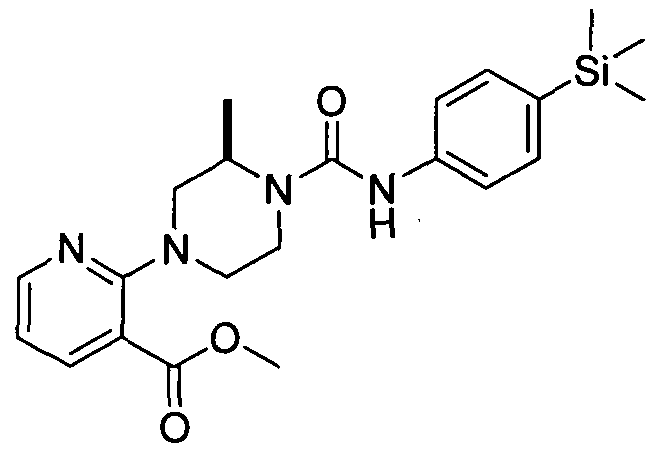 (/?)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate
(/?)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate
[0390]Synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine to give the title compound. MS:ES+ 427.1.1H NMR (400 MHz, DMSOd6) δ 8.26 (s, IH), 8.01 - 8.16 (m, IH), 7.66 - 7.81 (m, IH), 7.22 - 7.33 (m, 2H), 7.07 - 7.18 (m, 2H), 6.57 - 6.71 (m, IH), 4.14 - 4.27 (m, IH), 3.64 - 3.71 (m, IH), 3.37 - 3.52 (m, 2H), 3.01 - 3.07 (m, IH), 2.69 - 2.81 (m, IH), 2.16 - 2.36 (m, 4H), 0.85 - 0.99 (m, 3H), 0.00 (s, 9H)
[0391]2.35 Example 35 (Prepared according to Scheme 5, step a)
[0392]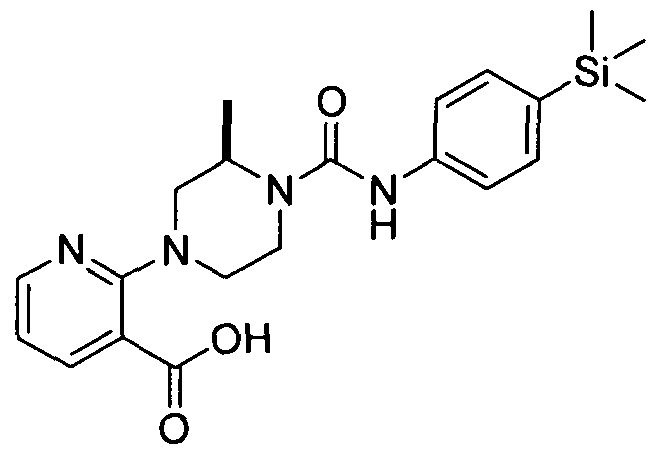
[0393](R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
[0394]Synthesised using a procedure similar to that described for Example 28 starting from (R)-Methyl 2-(3- methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate (Example 34). MS: ES+ 413.1.1H NMR (400 MHz, DMSOd6) δ 8.26 (s, IH), 8.04 - 8.11 (m, IH), 7.66 - 7.79 (m, IH), 7.21 - 7.31 (m, 2H), 7.07 - 7.21 (m, 2H), 6.59 - 6.69 (m, IH), 4.15 - 4.27 (m, IH), 3.63 - 3.72 (m, IH), 3.42 3.57 (m, 2H), 3.15 (br. s., IH), 2.97 - 3.05 (m, IH), 2.70 - 2.82 (m, IH), 0.91 - 0.99 (m, 3H), 0.00 (s, 9H)
[0395]2.36 Example 36 (Prepared according to Scheme 6, step a)
[0396]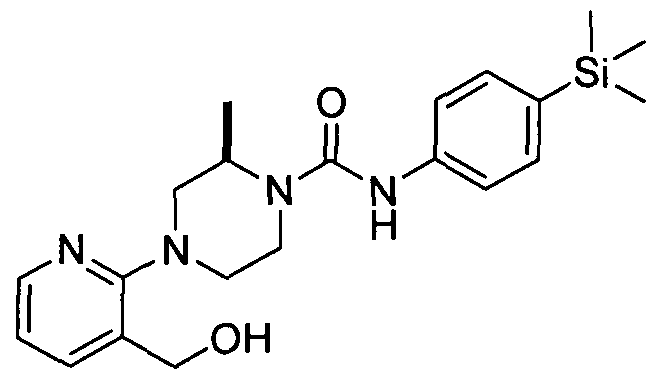
[0397](Λ)-4-(3-(Hydroxymethyl)pyridin-2-y])-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0398]Synthesised using a procedure similar to that described for Example 31 starting from (/?)-Methyl 2-(3- methyl-4-(4-(trimethylsilyl)ρhenylcarbamoyl)piperazin-l -yl)nicotinate (Example 34)
MS:ES+ 421.0.1H NMR (400 MHz, DMSO-d6) δ 8.29 (s, IH), 7.92 - 8.01 (m, IH), 7.52 - 7.63 (m, IH), 7.23 - 7.30 (m, 2H), 7.09 - 7.21 (m, 2H), 6.75 - 6.88 (m, IH), 5.00 - 5.12 (m, IH), 4.30 - 4.39 (m, 2H), 4.17 - 4.27 (m, IH), 3.71 - 3.81 (m, IH), 3.10 - 3.24 (m, 2H), 2.99 - 3.06 (m, IH), 2.69 (br. s., IH), 2.50 - 2.61 (m, IH), 1.07 - 1.15 (m, 3H), 0.00 (s, 9H)
[0399]2.37 Example 37 (Prepared according to Scheme 5 step b)
[0400]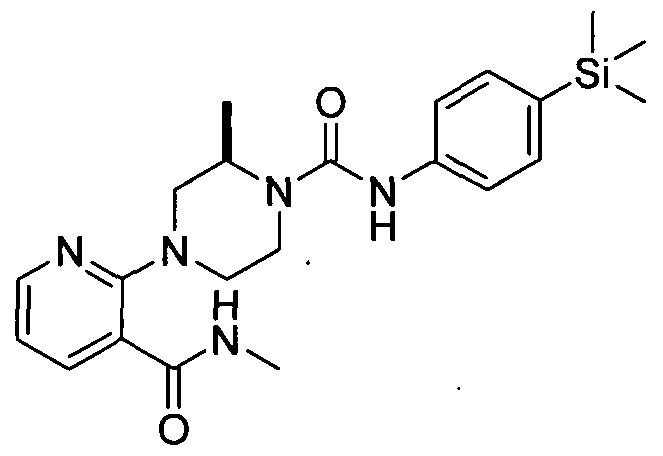 (R)-2-Methyl-4-(3-(methylcarbamoyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl) piperazine-1-carboxamide
(R)-2-Methyl-4-(3-(methylcarbamoyl)pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl) piperazine-1-carboxamide
[0401]Synthesised using a procedure similar to that described for Example 30 starting from (R)-2-(3-Methyl-4-(4-
[0402](trimethylsilyl)pheny lcarbamoyl)piperazin- 1 -yl)nicotinic acid
[0404]MS: ES+ 426.1.1H NMR (400 MHz, CDCl3) δ 8.12 - 8.21 (m, IH), 7.95 - 8.08 (m, 2H), 7.20 - 7.26 (m, 2H),
[0405]7.10 - 7.17 (m, 2H), 6.84 - 6.91 (m, IH), 6.14 (s, IH), 4.17 (br. s., IH), 3.69 - 3.79 (m, IH), 3.03 - 3.29 (m,
[0406]4H), 2.79 - 2.88 (m, 4H), 1.12 - 1.24 (m, 3H), 0.02 (s, 9H)
[0408]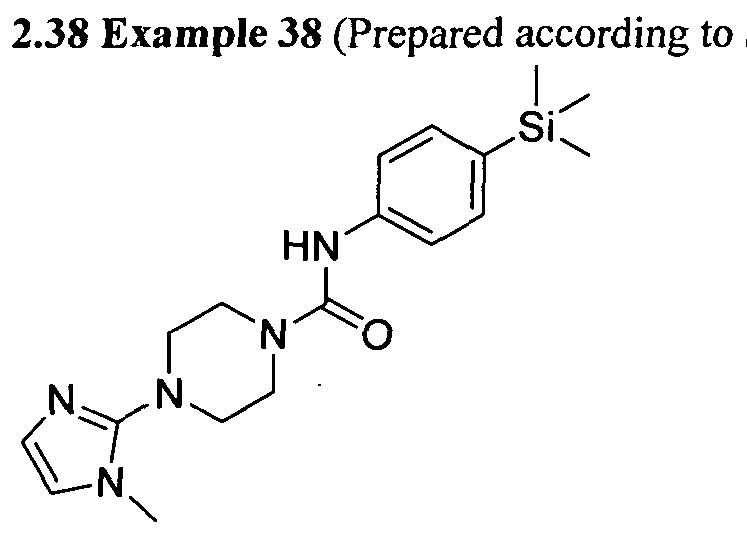
[0409]4-( 1 -Methyl- 1 H-imidazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 -carboxamide
[0410]The title compound was synthesised using a procedure similar to that described for Example 23 starting from
[0411]1 -(I -Methyl- 1 H-imidazol-2-yl)piperazine (EP0233051) and piperazine.
[0412]MS: ES+ 358.1H NMR (400 MHz, DMSO-J15) δ ppm 8.61 (s, 1 H) 7.43 - 7.50 (m, 2 H) 7.33 - 7.41 (m, 2 H)
[0413]6.86 - 6.92 (m, 1 H) 6.58 - 6.63 (m, 1 H) 3.53 - 3.62 (m, 4 H) 3.45 - 3.50 (m, 3 H) 2.91 - 3.01 (m, 4 H) 0.21 (s,
[0415]2.39 Example 39 (Prepared according to Scheme 4)
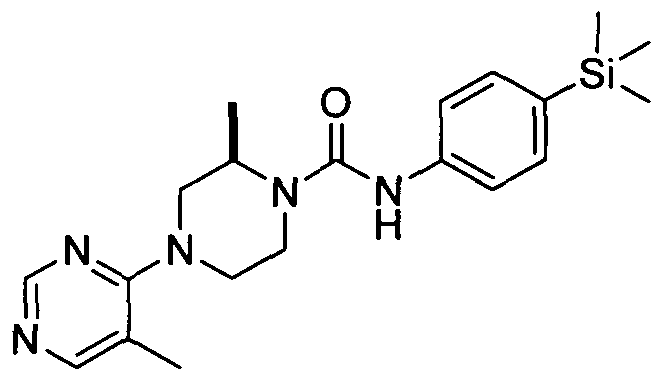
[0416](R)-2-Methyl-4-(5-methylpyrimidin-4-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide.
[0417]Synthesised in accordance with Example 23 using (i?)-(+)-2-methylpiperazine to give the title compound. MS: ES+ 384.2.1H NMR (400 MHz, CDCl3) δ 8.56 (s, IH), 8.04 (s, IH), 7.35 - 7.59 (m, 4H), 6.76 (s, IH), 4.42 - 4.54 (m, IH), 4.25 - 4.37 (m, IH), 3.99 - 4.14 (m, 2H), 3.53 - 3.64 (m, IH), 3.30 - 3.52 (m, 2H), 2.35 (s, 3H), 1.24 - 1.40 (m, 3H), 0.26 (s, 9H).
[0418]2.40 Example 40 (Prepared according to Scheme 4)
[0419]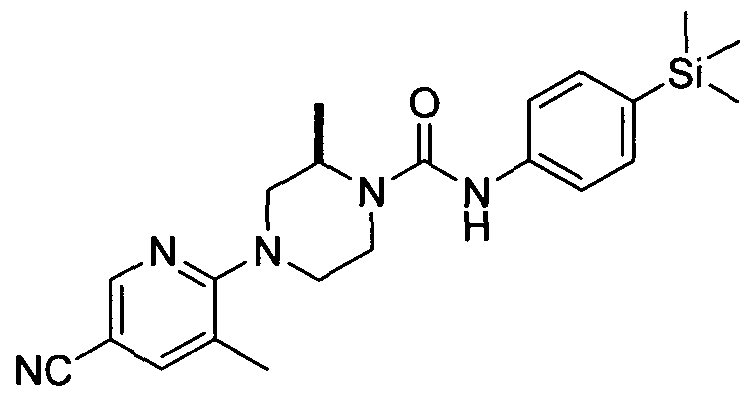 (R)-4-(5-Cyano-3-methylpyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
(R)-4-(5-Cyano-3-methylpyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0420]Synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine to give the title compound. MS: ES- 406.3.1H NMR (400 MHz, CDCl3) δ 8.36 - 8.51 (m, IH), 7.61 (s, IH), 7.43 - 7.53 (m, 2H), 7.33 - 7.43 (m, 2H), 6.40 (s, IH), 4.33 - 4.48 (m, IH), 3.88 - 4.02 (m, IH), 3.70 - 3.88 (m, IH), 3.55 - 3.67 (m, IH), 3.39 - 3.53 (m, IH), 3.22 - 3.33 (m, IH), 2.93 - 3.13 (m, IH), 2.36 (s, 3H), 1.33 - 1.43 (m, 3H), 0.27 (s, 9H)
[0421]2.41 Example 41 (Prepared according to Scheme 4)
[0422]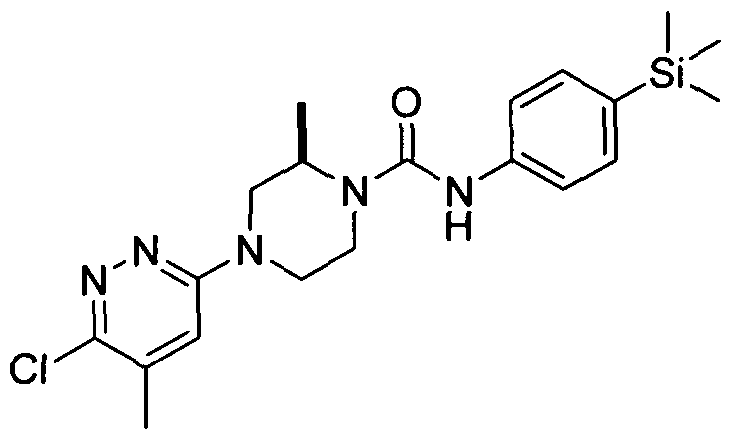
[0423](R)-4-(6-Chloro-5-methylpyridazin-3-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0424]Synthesised in accordance with Example 23 using (R)-(+)-2-methy]piperazine to give the title compound. MS: ES+ 418.1.1H NMR (400 MHz, DMSOd6) δ 8.32 (s, IH), 7.23 - 7.33 (m, 2H), 7.09 - 7.21 (m, 3H), 4.26 (br. s., I H), 4.00 - 4.10 (m, IH), 3.91 - 4.00 (m, 2H), 3.72 - 3.83 (m, 1 H), 2.92 - 2.99 (m, IH), 2.76 - 2.88 (m, IH), 2.06 (s, 3H), 0.87 - 0.98 (m, 3H), 0.00 (s, 9H)
2.42 Example 42 (Prepared according to Scheme 4)
[0425]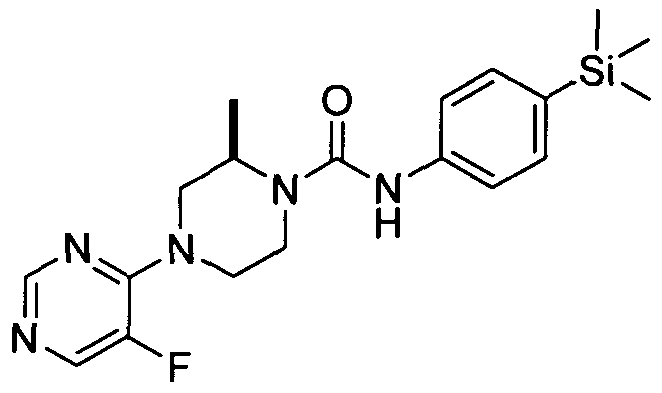
[0426](^^-(S-Fluoropyrimidin^-yO^-methyl-N^-CtrimethylsilyOphenyOpiperazine-l-carboxamide
[0427]Synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine to give the title compound.
[0428]MS:ES+ 388.2.1H NMR (400 MHz, DMSOd6) δ 8.34 (s, IH), 8.15 - 8.22 (m, IH), 8.05 - 8.1 1 (m, IH), 7.22 - 7.31 (m, 2H), 7.12 - 7.20 (m, 2H), 4.14 - 4.29 (m, 2H), 3.98 - 4.07 (m, IH), 3.71 - 3.81 (m, IH), 3.13 - 3.20 (m, IH), 2.88 - 3.08 (m, 2H), 0.87 - 0.96 (m, 3H), 0.00 (s, 9H)
[0429]2.43 Example 43 (Prepared according to Scheme 7)
[0430]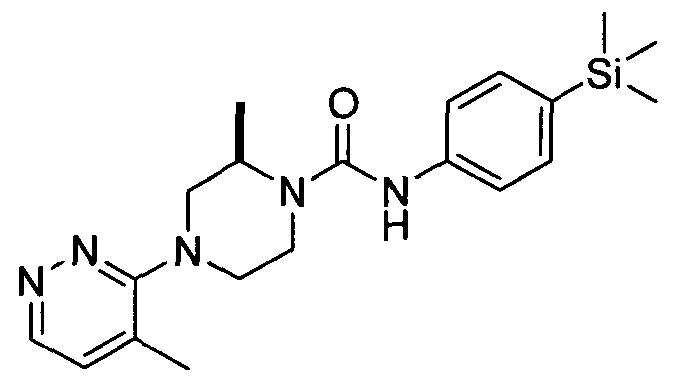
[0431](R)-2-Methyl-4-(4-methylpyridazin-3-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0432]A solution of (R)-4-(6-Chloro-4-methylpyridazin-3-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine- 1 - carboxamide
[0433](Example 32) (1.19 mmol) in EtOH was hydrogenated on an H-Cube hydrogenator reactor with 10% palladium on charcoal catalyst at 500C. The reaction was concentrated under reduced pressure. The resulting residue was purified by flash chromatography (0-10 % 2M ammonia in methanol in dichloromethane) yielding the title compound (0.37 mmol).
[0434]MS: ES+ 384.1.1H NMR (400 MHz, DMSOd6) δ 8.50 - 8.61 (m, IH), 8.34 (s, IH), 7.22 - 7.30 (m, 2H),
[0435]7.1 1 - 7.22 (m, 3H), 4.27 (br. s., IH), 3.73 - 3.85 (m, IH), 3.36 (br. s., IH), 3.18 - 3.28 (m, IH), 3.09 - 3.17
[0436](m, IH), 2.63 - 2.83 (m, 2H), 2.12 (s, 3H), 1.04 - 1.21 (m, 3H), 0.00 (s, 9H)
[0437]2.44 Example 44 (Prepared according to Scheme 8)
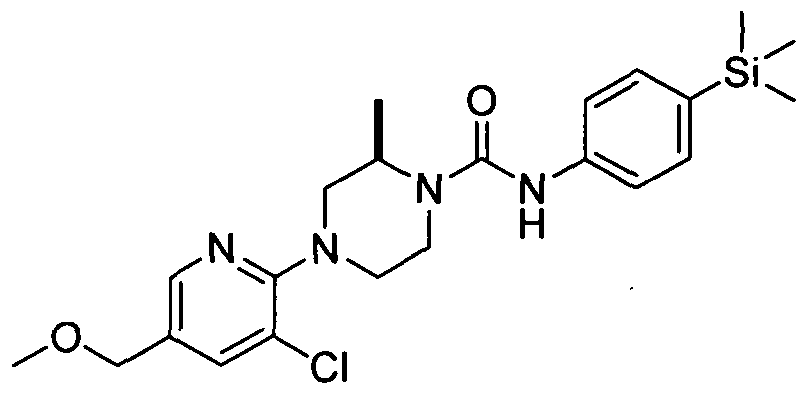
[0438](R)-4-(3-Chloro-5-(methoxymethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0439]To a solution of (5,6-dichloropyridin-3-yl)methanol (2.81 mmol) in THF (10 ml) was added NaH (2.81 mmmol) portionwise. The reaction was stirred at room temperature for 15 min. Methyl iodide (2.81 mmol) was added in a dropwise fashion and the reaction was stirred at room temperature for 16hrs. Water (10 ml) was added and the THF was removed under reduced pressure. The resulting residue was partitioned between DCM (50 ml) and water (50 ml). The organic phase was separated and dried using a phase separation cartridge and concentrated under reduced pressure. The resulting oil was purified by flash chromatography (0-20% ethyl acetate in petroleum ether) yielding the product 2,3-dichloro-5-(methoxymethyl) pyridine (1.85 mmol). 1H NMR (400 MHz, DMSOd6) δ 8.37 (br. s., IH), 8.09 (br. s., IH), 4.48 (s, 2H), 3.19 - 3.46 (m, 3H)
[0440]The title compound was synthesised in accordance with Example 23 using (i?)-(+)-2-methylpiperazine and the intermediate, 2,3-dichloro-5-(methoxymethyl) pyridine, described above.
[0441]MS: ES+ 447.04.1H NMR (400 MHz, CDCl3) δ 8.15 (br. s., IH), 7.67 (br. s., IH), 7.44 - 7.55 (m, 2H), 7.36 - 7.44 (m, 2H), 6.39 (br. s., IH), 4.40 (br. s., 3H), 3.89 - 4.03 (m, IH), 3.71 - 3.89 (m, 2H), 3.33 - 3.59 (m, 4H), 2.89 - 3.17 (m, 2H), 1.41 - 1.52 (m, 3H), 0.21 - 0.34 (m, 9H)
[0442]2.45 Example 45 (Prepared according to Scheme 7)
[0443]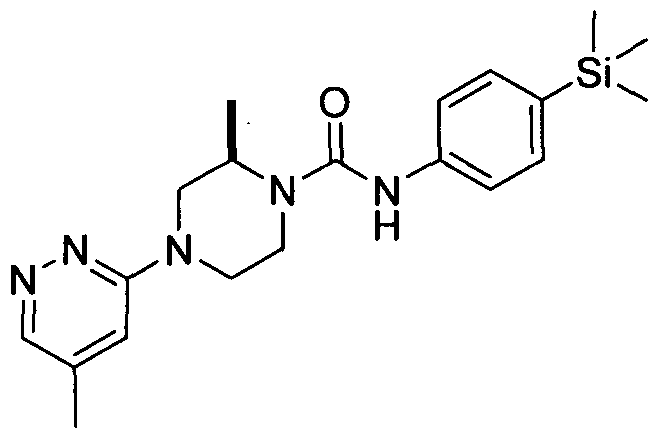
[0444](/?)-2-Methyl-4-(5-methylpyridazin-3-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0445]Synthesised using a procedure similar to that described for Example 43 starting from (R)-4-(6-Chloro-5- methylpyridazin-3-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide (Example 41).
MS: ES+ 384.1.1H NMR (400 MHz, DMSOd6) δ 8.31 (br. s., IH), 8.22 (br. s., IH), 7.20 - 7.36 (m, 2H), 7.07 - 7.20 (m, 2H), 6.91 (br. s., IH), 4.25 (br. s., IH), 4.07 (br. s., IH), 3.97 (br. s., IH), 3.76 (br. s., IH), 2.91 - 3.04 (m, 2H), 2.65 - 2.85 (m, IH), 2.01 (br. s., 3H), 0.83 - 1.02 (m, 3H), 0.00 (br. s., 9H)
[0446]2.46 Example 46 (Prepared according to Scheme 5)
[0447]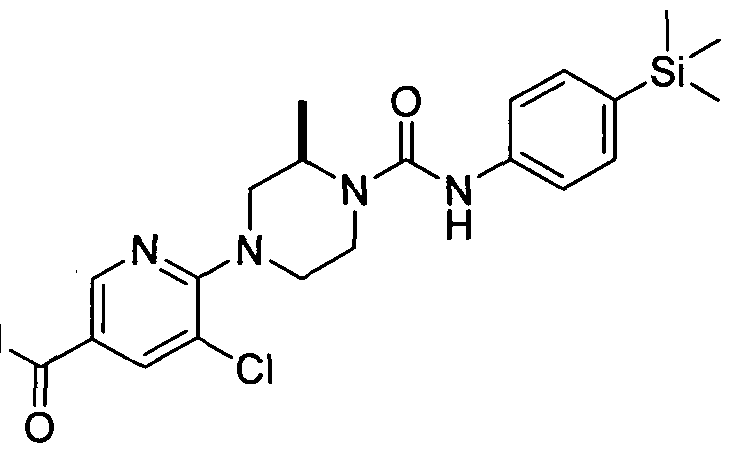
[0448](R)-4-(3-Chloro-5-(dimethylcarbamoyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0449]Synthesised using a procedure similar to that described for Example 30 starting from (R)-5-Chloro-6-(3- methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid (Example 28). MS: ES+ 474.1.1H NMR (400 MHz, DMSOd6) δ 8.32 (s, IH), 8.02 - 8.12 (m, IH), 7.63 - 7.71 (m, IH), 7.22 - 7.32 (m, 2H), 7.12 - 7.19 (m, 2H), 4.23 - 4.30 (m, IH), 3.74 - 3.87 (m, IH), 3.55 - 3.67 (m, 2H), 2.61 2.89 (m, 9H), 1.01 - 1.11 (m, 3H), 0.00 (s, 9H)
[0450]2.47 Example 47 (Prepared according to Scheme 4)
[0451]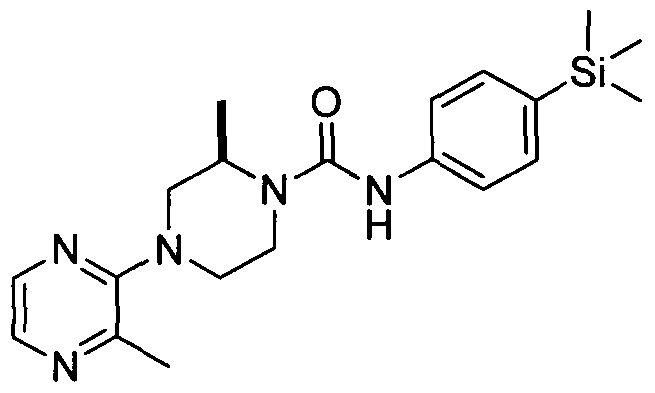
[0452](R)-2-Methyl-4-(3-methylpyrazin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0453]Synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine to give the title compound. MS: ES+ 384.1.1H NMR (400 MHz, DMSOd6) δ 8.33 (s, IH), 7.85 - 7.94 (m, 2H), 7.22 - 7.31 (m, 2H), 7.13 - 7.19 (m, 2H), 4.20 - 4.29 (m, IH), 3.75 - 3.84 (m, IH), 3.27 - 3.36 (m, IH), 3.20 - 3.27 (m, IH), 3.02 3.07 (m, IH), 2.65 - 2.73 (m, IH), 2.53 - 2.63 (m, IH), 2.31 (s, 3H), 0.96 - 1.25 (m, 3H), 0.00 (s, 9H)
[0454]2.48 Example 48 (Prepared according to Scheme 8)
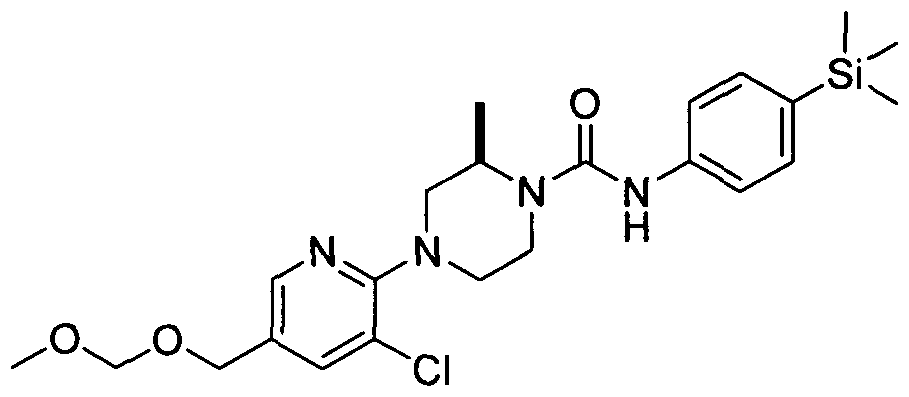
[0455](/?)-4-(3-Chloro-5-((methoxymethoxy)methyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine- 1-carboxamide
[0456]Synthesised in accordance with Example 44 using (R)-(+)-2-methylpiperazine to give the title compound. MS:ES+ 477.1.1H NMR (400 MHz, CDCl3) δ 8.01 - 8.10 (m, IH), 7.53 - 7.58 (m, IH), 7.31 - 7.39 (m, 2H), 7.24 - 7.31 (m, 2H), 6.25 (s, IH), 4.60 (s, 2H), 4.42 (s, 2H), 4.22 - 4.33 (m, IH), 3.78 - 3.87 (m, IH), 3.62 - 3.76 (m, 2H), 3.33 - 3.43 (m, IH), 3.31 (s, 3H), 2.96 - 3.04 (m, IH), 2.83 - 2.94 (m, IH), 1.26 - 1.40 (m, 3H), 0.15 (s, 9H)
[0457]2.49 Example 49 (Prepared according to Scheme 9)
[0458]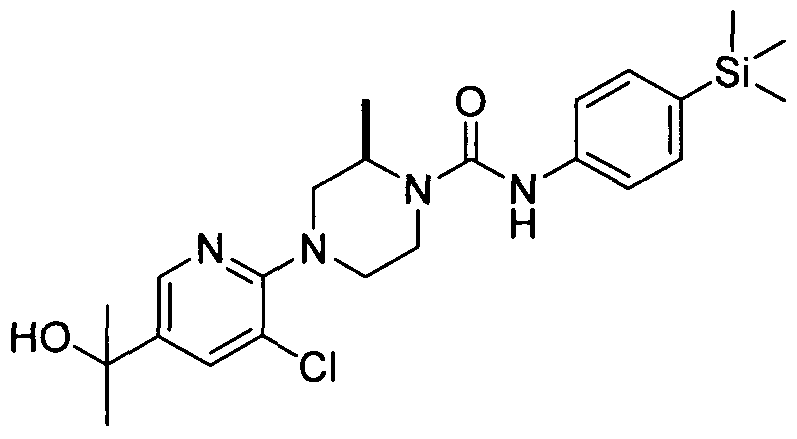
[0459](R)-4-(3-Chloro-5-(2-hydroxypropan-2-yl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsiIyl)phenyI)piperazine-l- carboxamide
[0460]To a solution of ethyl 5,6-dichloronicotinate (2.27mmol) in THF (15 ml) at -780C was added methyl magnesium bromide (3M in THF, 11.4mmol) in a dropwise fashion. The reaction was stirred at -780C for 2 hours. The reaction was quenched with saturated ammonium chloride (10 ml) and extracted with ethyl acetate (50 ml). The organic phase was separated, dried with a phase separation cartridge and concentrated under reduced pressure. The resulting oil was purified by flash chromatography (0-20% ethyl acetate in petroleum ether) yielding the product 2-(5,6-dichloropyridin-3-yl)propan-2-ol (2.18mmol).
[0461]MS: ES+ 205.9.1H NMR (400 MHz, CDCl3) δ 8.23 - 8.40 (m, IH), 7.79 - 8.00 (m, IH), 1.78 (br. s., IH), 1.54 (s, 6H)
[0462]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine and the above intermediate 2-(5,6-dichloropyridin-3-yl)propan-2-ol.
[0463]MS: ES+ 461.2.1H NMR (400 MHz, CDCI3) δ 8.28 - 8.33 (m, IH), 7.78 - 7.83 (m, IH), 7.44 - 7.52 (m, 2H), 7.36 - 7.43 (m, 2H), 6.38 (s, IH), 4.32 - 4.44 (m, IH), 3.89 - 4.00 (m, IH), 3.71 - 3.85 (m, 2H), 3.43 - 3.54 (m, IH), 3.05 - 3.14 (m, IH), 2.93 - 3.04 (m, IH), 1.77 (s, IH), 1.61 (s, 6H), 1.44 - 1.51 (m, 3H), 0.27 (s, 9H)
2.50 Example 50 (Prepared according to Scheme 10)
[0464]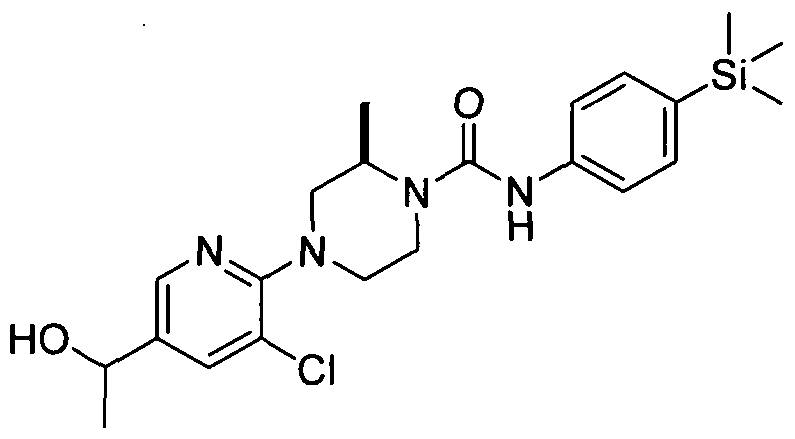
[0465](2R)-4-(3-Chloro-5-(l-hydroxyethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0466]A solution of (5,6-dichloropyridin-3-yl) methanol (5.62mmol) and manganese dioxide (52mmol) in DCM (50 ml) was stirred at room temperature for 4 days. The reaction was filtered through celite, washed with EtOH and concentrated under reduced pressure yielding the crude product which was used without purification, 5,6- dichloronictoninaldehyde. To a solution of the crude 5,6-dichloronictoninaldehyde (3.24mmol) in THF (20ml) at -780C was added methyl magnesium bromide (9.72mmol) in a dropwise fashion. The reaction was allowed to warm to room temperature overnight. The reaction was quenched with saturated ammonium chloride (50ml) and extracted with ethyl acetate (50 ml) The organic phase was separated, dried with a phase separation cartridge and concentrated under reduced pressure yielding the crude oil which was purified by flash chromatography (0-30% ethyl acetate in petroleum ether) yielding the product. l-(5,6-dichloropyridin- 3-yl)ethanol (2.14mmol).
[0467]MS: ES+ 191.9.1H NMR (400 MHz, CDCl3) δ 8.29 (s, IH), 7.75 - 8.03 (m, IH), 4.92 - 5.11 (m, IH), 2.11 - 2.26 (m, IH), 1.45 - 1.56 (m, 3H)
[0468]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine and the above intermediate l-(5,6-dichloropyridin-3-yl)ethanol.
[0469]MS: ES+ 447.2.1H NMR (400 MHz, CDCl3) δ 8.07 (s, IH), 7.60 (t, IH), 7.32 - 7.40 (m, 2H), 7.24 - 7.31 (m, 2H), 6.26 (s, IH), 4.74 - 4.85 (m, IH), 4.21 - 4.32 (m, IH), 3.76 - 3.88 (m, IH), 3.59 - 3.74 (m, 2H), 3.30 - 3.43 (m, IH), 2.93 - 3.04 (m, IH), 2.80 - 2.93 (m, IH), 1.71 - 1.77 (m, IH), 1.38 - 1.44 (m, 3H), 1.32 - 1.38 (m, 3H), 0.15 (s, 9H)
[0470]2.51 Example 51 (Prepared according to Scheme 1 1)
[0471]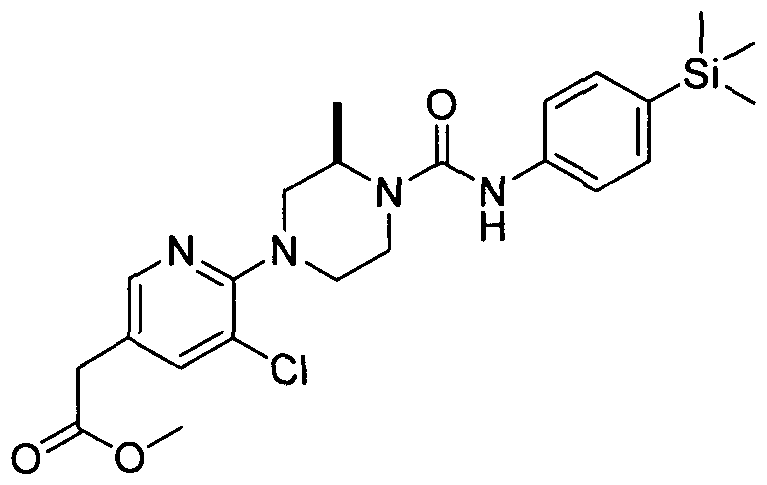 (R)-Methyl 2-(5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridin-3- yl)acetate
(R)-Methyl 2-(5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridin-3- yl)acetate
[0472]To a solution of (5,6-dichloropyridin-3-yl) methanol (28mmol) and TEA (56mmol) in DCM (250 ml) at 00C was added MsCl (28mmol) in a dropwise fashion. The reaction was stirred at room temperature for 2 hours. The reaction mixture was washed with saturated sodium bicarbonate (250 ml) and the organic was separated and dried with a phase separation cartridge and concentrated under reduced pressure yielding the product, (5,6-dichloropyridin-3-yl)methyl methanesulfonate (22.4mmol). MS: ES+ 255.9.1H NMR (400 MHz, CDCl3) δ 8.36 (s, IH), 7.90 (s, IH), 5.24 (s, 2H), 3.09 (s, 3H)
[0473]To a solution of (5,6-dichloropyridin-3-yl)methyl methanesulfonate (22.4mmol) and TEA (22.4mmol) in THF (80 ml) and water (10 ml) was added sodium cyanide portionwise under nitrogen. The reaction was stirred under nitrogen for 2 hours. The reaction was diluted with saturated ammonium chloride (250 ml) and extracted with ethyl acetate (2 x 250 ml). The organic layer was separated, dried over magnesium sulphate and concentrated under reduced pressure yielding the crude oil. This was purified by flash chromatography (0-25 % ethyl acetate in petroleum ether) yielding the product, 2-(5,6-dichloropyridin-3-yl)acetonitrile (13.1mmol). MS: ES" 184.9.1H NMR (400 MHz, DMSOd6) δ 8.36 - 8.51 (m, IH), 8.08 - 8.25 (m, IH), 4.14 (s, 2H)
[0474]A solution of 2-(5,6-dichloropyridin-3-yl)acetonitrile (12.8mmol) in 2M sodium hydroxide (64.2mmol) was heated in the microwave at 100 °C for 20 minutes. The reaction was washed with ethyl acetate (20 ml). The aqueous was then acidified to pH 1 with 2N hydrochloric acid and extracted with ethyl acetate (2 x 30 ml) the organics were combined, dried with a phase separation cartridge and concentrated under reduced pressure yielding the product, 2-(5,6-dichloropyridin-3-yl) acetic acid (12.8mmol).
[0475]MS: ES+ 205.9.1H NMR (400 MHz, DMSOd6) δ 12.29 (br. s., IH), 8.18 - 8.53 (m, IH), 7.90 - 8.18 (m, IH), 3.71 (s, 2H).
[0476]A solution of 2-(5,6-dichloropyridin-3-yl) acetic acid (12.8mmol) in methanol (40 ml) and concentrated sulphuric acid (10 ml) was stirred at room temperature for 2 hours. The methanol was removed under reduced pressure and the residue was partitioned between ethyl acetate (50 ml) and water (50 ml). The organic phase was separated, dried with a phase separator cartridge and concentrated under reduced pressure yielding a crude oil. The resultant oil was purified by flash chromatography (0-25 % ethyl acetate in petroleum ether) yielding the product, methyl 2-(5,6-dichloropyridin-3-yl acetate (8.96mmol).
[0477]MS: ES+ 219.9.1H NMR (400 MHz, DMSOd6) δ 8.28 - 8.39 (m, IH), 8.07 - 8.14 (m, IH), 3.83 (s, 2H), 3.66 (s, 3H).
[0478]The title compound was synthesised from the above intermediate methyl 2-(5,6-dichloropyridin-3-yl acetate and (/?)-(+)-2-methylpiperazine in a similar procedure to that described for Example 23.
MS: ES+ 475.2.1H NMR (400 MHz, CDCl3) δ 7.82 - 7.87 (m, IH), 7.35 - 7.40 (m, IH), 7.19 - 7.25 (m, 2H), 7.10 - 7.18 (m, 2H), 6.11 (s, IH), 4.13 (br. s., IH), 3.64 - 3.74 (m, IH), 3.52 - 3.62 (m, 2H), 3.50 (s, 3H), 3.19 - 3.29 (m, IH), 2.80 - 2.93 (m, IH), 2.66 - 2.80 (m, IH), 1.31 (s, 2H), 1.19 - 1.25 (m, 3H), 0.02 (s, 9H).
[0479]2.52 Example 52 (Prepared according to Scheme 11, step g)
[0480]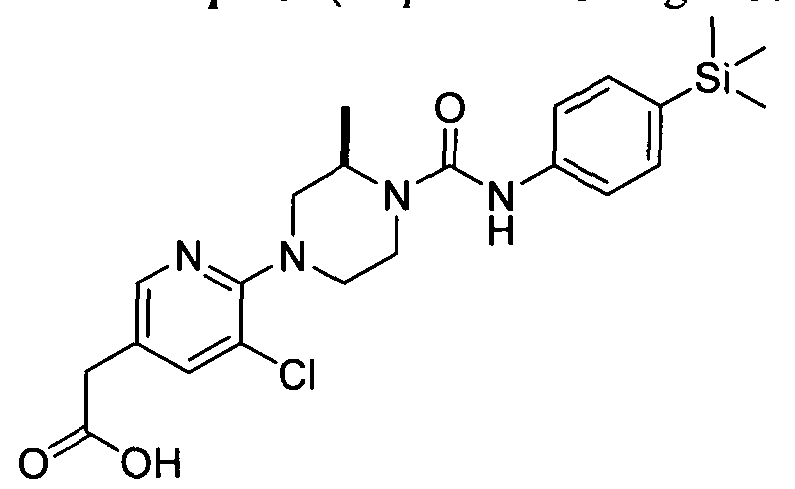 R)-2-(5-Chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridin-3-yl)acetic acid
R)-2-(5-Chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridin-3-yl)acetic acid
[0481]A solution of (R)-methyl 2-(5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)-piperazin-l - yl)pyridin-3-yl)acetate (Example 51) (0.42mmol) and potassium hydroxide (1.26mmol) in THF (9 ml) and water (3 ml) was stirred at room temperature for 3 hrs. The reaction was concentrated under reduced pressure. The resulting residue was diluted with water (20 ml) and acidified with 2N hydrochloric acid to pH 1. The resultant suspension was extracted with ethyl acetate. The organic phase was separated, dried with a phase separation cartridge and concentrated under reduced pressure to yield the product after trituration with DCM/petroleum ether (1 :1). (R)-2-(5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l- yI)pyridin-3-yl)acetic acid (0.21mmol).
[0482]MS: ES+ 461.5.1H NMR (400 MHz, CDCl3) δ 8.08 - 8.14 (m, IH), 7.60 - 7.66 (m, IH), 7.43 - 7.52 (m, 2H), 7.36 - 7.42 (m, 2H), 6.44 (s, IH), 4.31 - 4.45 (m, IH), 3.89 - 3.99 (m, IH), 3.70 - 3.87 (m, 2H), 3.61 (s, 2H), 3.41 - 3.54 (m, IH), 3.05 - 3.15 (m, IH), 2.92 - 3.03 (m, IH), 1.41 - 1.49 (m, 3H), 0.27 (s, 9H)
[0483]2.53 Example 53 (Prepared according to Scheme 12)
[0484]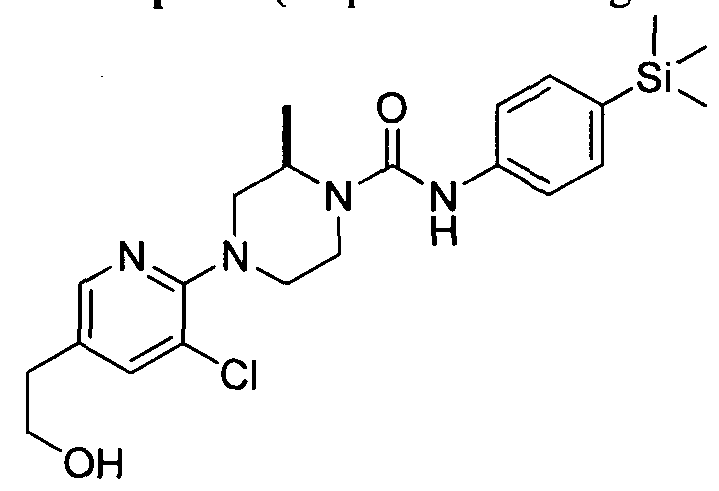
[0485](R)-4-(3-Chloro-5-(2-hydroxyethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0486]To a solution of (R)-methyl 2-(5-chloro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)-piperazin-l- yl)pyridin-3-yl)acetate (Example 51) (0.67mmol) in THF (10 ml) was added lithium aluminium hydride (0.808mmol) at 00C. The reaction was stirred at 00C for 1 hr. The reaction was quenched with sodium hydroxide (2M, 5ml) and water (15 ml). The solution was then extracted with ethyl acetate (20 x 2). The
organics were combined, dried (MgSO,}) and concentrated under reduced pressure yielding (R)-4-(3-chloro-5- (2-hydroxyethyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide (0.33mmol). MS: ES+ 447.2.1H NMR (400 MHz, CDCl3) δ 8.06 - 8.12 (m, IH), 7.54 - 7.59 (m, IH), 7.44 - 7.51 (m, 2H), 7.36 - 7.42 (m, 2H), 6.38 (s, IH), 4.31 - 4.44 (m, IH), 3.82 - 3.99 (m, 3H), 3.69 - 3.82 (m, 2H), 3.40 - 3.54 (m, IH), 3.04 - 3.13 (m, IH), 2.93 - 3.03 (m, IH), 2.76 - 2.85 (m, 2H), 1.57 (s, IH), 1.46 - 1.49 (m, 3H), 0.27 (s, 9H).
[0487]2.54 Example 54 (Prepared according to Scheme 4)
[0488]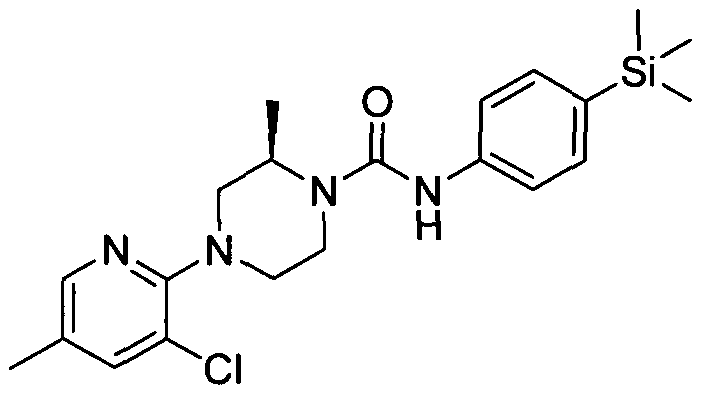
[0489](R)-4-(3-chloro-5-methylpyridin-2-yl)-2-methyl-N-(4
[0490](trimethylsilyθphenyl)piperazine-l-carboxamide
[0491]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine.
[0492]MS: ES+ 415.2.1H NMR (400MHz, MeOD ) δ 0 (s, 9H), 1.2 (d, 3H), 2 (s, 3H), 2.65 (t, IH), 2.75 (d, IH), 3
[0493](s, IH), 3.4 (t, 2H), 3.8 (d, IH), 4.3 (br.s, IH), 7.1 (d, 2H), 7.2 (d, 2H), 7.4 (s, IH), 7.8 (s, IH).
[0494]2.55 Example 55 (Prepared according to Scheme 4)
[0495]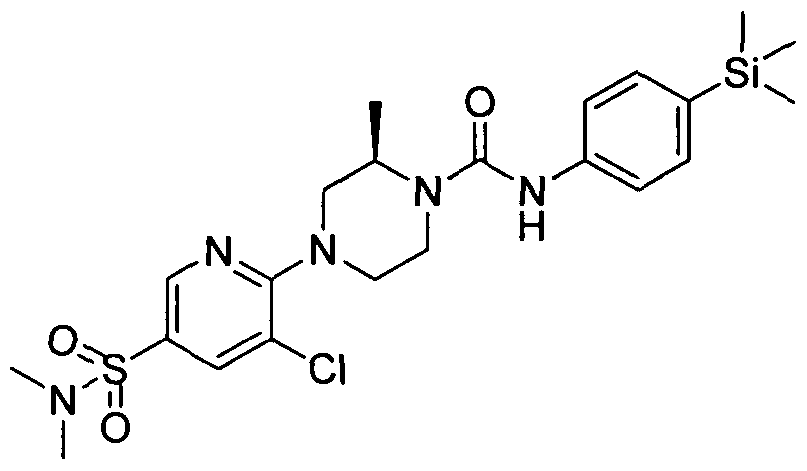
[0496](R)-4-(3-chloro-5-(N,N-dimethylsulfamoyl)pyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide.
[0497]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine and
[0498]5,6-dichloro-N,N-dimethylpyridine-3-sulfonamide [CAS622339-79-l].
[0499]MS: ES+ 510.1.1H NMR (400 MHz, DMSO) δ 0 (s, 9H), 1 (d, 3H), 2.45 (s, 6H), 2.8 (t, IH), 2.95 (d, IH),
[0500]3.8 (t, 3H), 4.3 (bs, IH), 7.1 (d, 2H), 7.25 (d, 2H), 7.8 (s, IH), 8.3 (d, 2H).
[0501]2.56 Example 56 (Prepared according to Scheme 4)
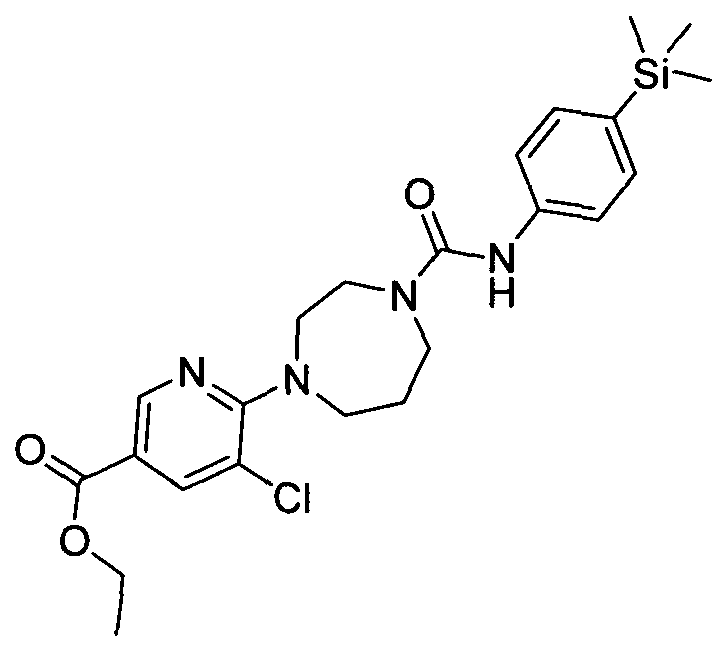
[0502]Ethyl 5-chloro-6-(4-(4-(trimethy lsilyOpheny lcarbamoyl)- 1 ,4-diazepan- 1 -y l)nicotinate
[0503]The title compound was synthesised using a procedure similar to that used for Example 23 and using homopiperazine.
[0504]MS: ES+ 447.2.1H NMR (400 MHz, CDCl3) δ 8.62 - 8.85 (m, IH), 8.02 - 8.23 (m, IH), 7.44 - 7.57 (m, 2H),
[0505]7.32 - 7.44 (m, 2H), 6.38 (s, IH), 4.26 - 4.49 (m, 2H), 3.89 - 4.21 (m, 4H), 3.74 - 3.89 (m, 2H), 3.50 - 3.68 (m,
[0506]2H), 1.98 - 2.24 (m, 2H), 1.27 - 1.47 (m, 3H), 0.26 (s, 9H)
[0507]2.57 Example 57 (Prepared according to Scheme 9)
[0508]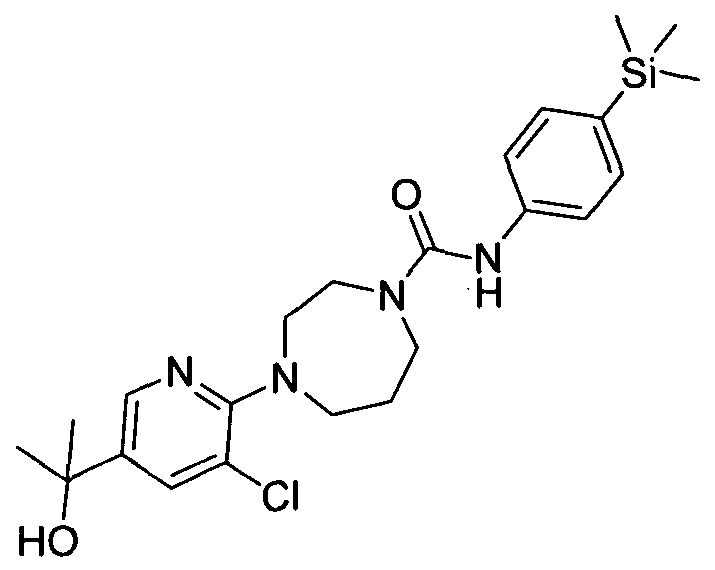
[0509]4-(3 -Chloro-5-(2-hydroxypropan-2-y i)pyridin-2-yl)-N-(4-(trimethylsi Iy I)pheny I)- 1 ,4-diazepane- 1 - carboxamide
[0510]The title compound was synthesised using homopiperazine and 2-(5,6-dichloropyridin-3-yl)propan-2-ol in accordance with Example 49.
[0511]MS: ES+ 461.2.1H NMR (400 MHz, CDCl3) δ 8.08 - 8.31 (m, IH), 7.66 - 7.80 (m, IH), 7.41 - 7.58 (m, 2H), 7.35 - 7.41 (m, 2H), 6.44 (s, IH), 3.70 - 3.90 (m, 6H), 3.57 - 3.70 (m, 2H), 1.94 - 2.19 (m, 2H), 1.67 (s, IH),
[0512]1.57 (s, 6H), 0.26 (s, 9H)
[0513]2.58 Example 58 (Prepared according to Scheme 5, step a)
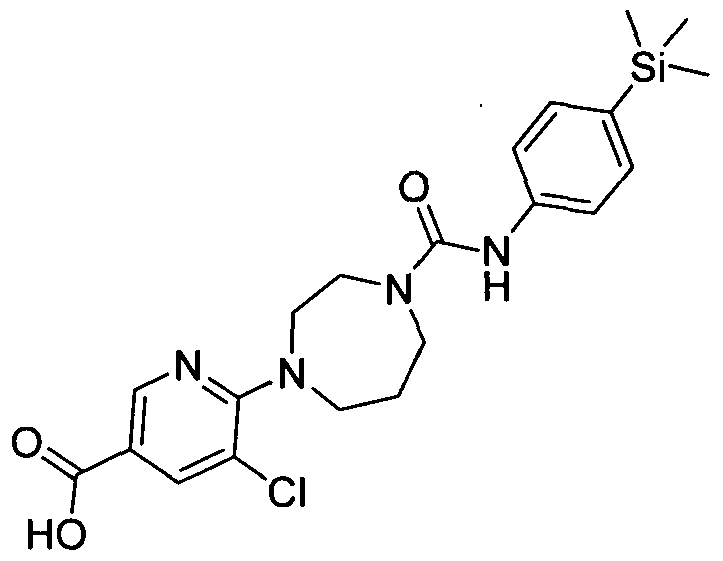 5-Chloro-6-(4-(4-(trimethy]silyl)phenylcarbamoyl)-l,4-diazepan-]-yl)nicotinic acid
5-Chloro-6-(4-(4-(trimethy]silyl)phenylcarbamoyl)-l,4-diazepan-]-yl)nicotinic acid
[0514]The title compound was synthesised in accordance with Example 28 using Ethyl 5-chloro-6-(4-(4- (trimethylsilyl)phenylcarbamoyl)-l,4-diazepan-l-yl)nicotinate (Example 56) as starting material. MS: ES+ 447.1.1H NMR (400 MHz, DMSOd6) δ 12.55 - 12.86 (m, IH), 8.36 (s, IH), 8.10 (s, IH), 7.77 (s, IH), 7.04 - 7.28 (m, 4H), 3.70 (br. s., 2H), 3.56 - 3.65 (m, 2H), 3.51 (br. s., 2H), 3.33 (br. s., 2H), 1.74 (br. s., 2H), 0.00 (s, 9H).
[0515]2.59 Example 59 (Prepared according to Scheme 4)
[0516]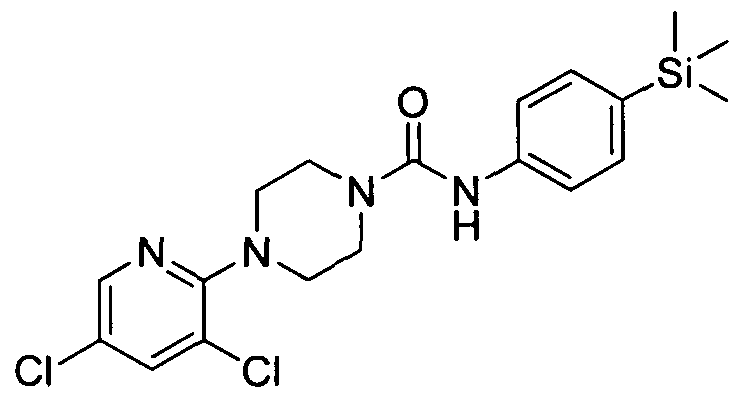
[0517]4-(3,5-dichloropyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0518]Synthesised in accordance with Example 23 using piperazine to give the title compound.
[0519]MS: ES+ 423.21.1H NMR (400 MHz, CDCl3) δ 8.15(1H, s), 7.65(1H, s), 7.45(2H, d), 7.35(2H, d), 6.35(1H, s), 3.7(4H, d), 3.4(4H, d), 0.3(9H, s)
[0520]2.60 Example 60 (Prepared according to Scheme 13)
[0521]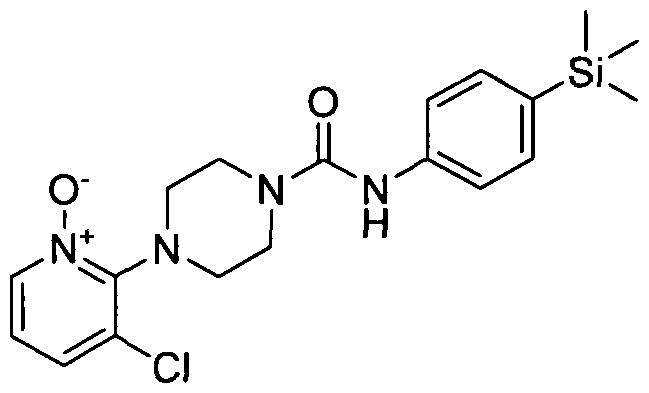 3-Chloro-2-(4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l -yl)pyridine 1 -oxide
3-Chloro-2-(4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l -yl)pyridine 1 -oxide
[0522]Solid sodium bicarbonate (0.15 mmol) was added to a solution of4-(3-Chloropyridin-2-yl)-N-(4- (trimethylsilyl)phenyl)piperazine-l-carboxamide (Example 13) (0.051 mmol) in DCM (5 ml). The mixture was then cooled in an ice-bath and treated with mCPBA (0.051 mmol). Stirring was continued, allowing the
reaction to slowly warm to room temperature overnight. The reaction was then re-cooled in ice, and treated with more sodium bicarbonate (0.15mmol) and raCPBA (0.051mmol), and again allowed to slowly warm to room temperature over 24 h. After re-cooling in ice, further mCPBA (0.051mmol) was added. The reaction was stirred for a further 24 h, then diluted with ethyl acetate (20 ml) and washed with saturated sodium bicarbonate (20 ml), 0.5 M Na2CO3 (20 ml), 5% Na2S2O5 solution (20 ml), saturated brine (20 ml), dried over Na2SO4 and evaporated to leave an amber film. This crude product was purified by flash chromatography (3- 15% methanol in DCM) yielding the title compound (0.019mmol).
[0523]ES+ 405, 407.1H NMR (400 MHz, CD3OD) δ 8.22 (dd, 1 H), 7.95 (dd, 1 H), 7.37 (dd, 1 H), 7.08 - 7.28 (m, 4 H), 3.83 - 4.12 (m, 4 H), 3.62 - 3.82 (m, 4 H), 0.01 (s, 9 H).
[0524]2.61 Example 61 (Prepared according to Scheme 14)
[0525]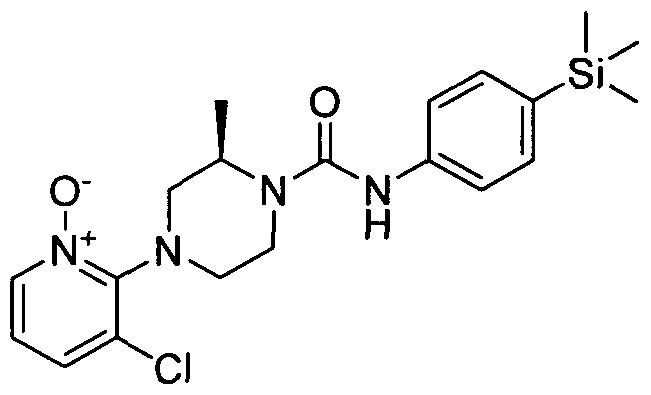
[0526](R)-3-Chloro-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyridine 1 -oxide
[0527]2,3-Dichloropyridine 1-oxide (CAS 53976-65-1) (l .l δmmol), (R)-2-methylpiperazine (l.lδmmol) and MP- Carbonate (2.36mmol) were combined in propan-2-ol (5 ml) in a microwave tube, sealed and heated in the microwave to 1500C for 90 minutes. The reaction mixture was cooled, filtered and evaporated to leave a brown gum (252 mg), which was determined to be a mixture of (R)-3-chloro-2-(3-methylpiperazin-l- yl)pyridine 1-oxide (67%) and starting 2,3-dichloropyridine 1-oxide (33%).
[0528]The crude intermediate (239 mg) was then combined with Intermediate 1 (0.77mmol), TEA (lmmol) and 4- dimethylaminopyridine (catalytic) in EtOH (4 ml), sealed in a microwave vessel and heated to 1000C, for 30 minutes. The reaction mixture was cooled and evaporated and the residue partitioned between ethyl acetate (30 ml) and 0.5 M Na2CO3 (20 ml). The organic layer was washed with water (10 ml), dilute citric acid (10 ml), water (10 ml), saturated sodium bicarbonate (10 ml), brine (20 ml), dried over Na2SO4 and evaporated to leave an amber gum. This crude product was then purified by flash chromatography (2-7% MeOH/DCM) and the combined product fractions freeze-dried yielding the title compound as a white solid (0.25mmol). ES+ 419, 421.1H NMR (400 MHz, CDCl3) δ 8.09 (d, 1 H), 7.41 - 7.52 (m, 2 H), 7.33 - 7.41 (m, 2 H), 7.28 - 7.33 (m, 1 H), 6.84 - 7.03 (m, 1 H), 6.41 (s, 1 H), 4.33 (br. s., 1 H), 2.90 - 4.10 (m, 6 H), 1.43 (d, 3 H), 0.25 (s, 9 H).
[0529]2.62 Example 62 (Prepared according to Scheme 4)
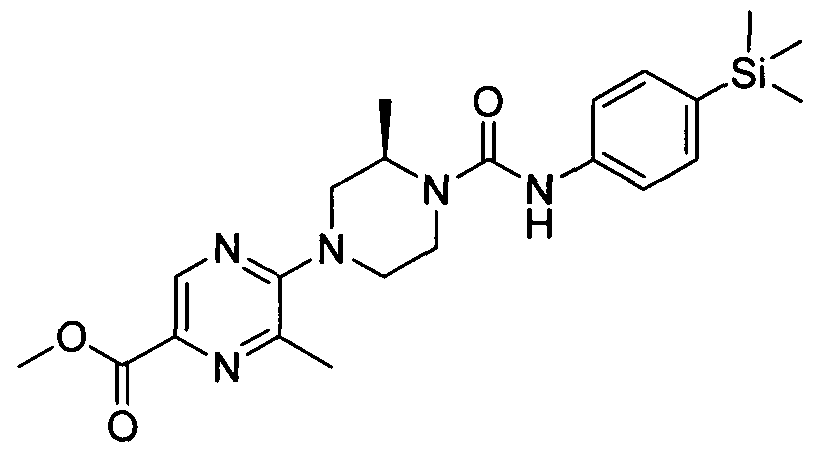
[0530](R)-Methyl 6-methyl-5-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyrazine-2- carboxylate
[0531]The title compound was synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine and methyl S-chloro-ό-methylpyrazine^-carboxylate (CAS 77168-85-5).
[0532]MS: ES+ 442.1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1 H), 7.42 - 7.51 (m, 2 H), 7.33 - 7.41 (m, 2 H), 6.38 (s, 1 H), 4.31 - 4.53 (m, 1 H), 3.83 - 4.08 (m, 5 H), 3.72 (m, 1 H), 3.48 (m, 1 H), 3.30 (m, 1 H), 3.10 (m, 1 H), 2.65 (s, 3 H), 1.38 (d, J=6.82 Hz, 3 H), 0.25 (s, 9 H).
[0533]2.63 Example 63 (Prepared according to Scheme 4)
[0534]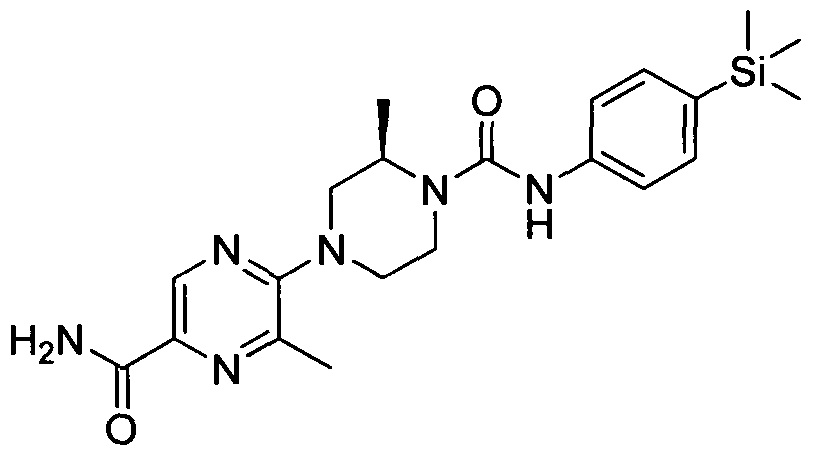
[0535](R)-6-Methyl-5-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)pyrazine-2-carboxamide
[0536](R)-Methyl 6-methyl-5-(3-methylpiperazin-l-yl)pyrazine-2-carboxylate (0.33mmol) (prepared in Example 62) was dissolved in ammonia (2.0M in MeOH, 20 ml) and left to stand in a stoppered flask at room temperature for 4 days. The solvent was evaporated to yield (R)-6-methyl-5-(3-methylpiperazin-l- yl)pyrazine-2-carboxamide (0.33mmol).
[0537]MS: ES+ 236.1H NMR (400 MHz, DMSOd6) δ 8.57 (s, 1 H), 7.77 (br. s., 1 H), 7.48 (br. s., 1 H) 3.49 - 3.77 (m, 2 H), 2.76 - 2.96 (m, 4 H), 2.43 - 2.53 (m, 1 H, under DMSO), 2.15 - 2.41 (m, 1 H) 1.01 (d, J=6.06 Hz, 3 H).
[0538]The title compound was synthesised in accordance with Example 23 using (/?)-(+)-2-methylpiperazine and (Λ)-methyl 6-methyl-5-(3-methylpiperazin-l-yl)pyrazine-2 -carboxylate prepared above. MS: ES+ 427.1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1 H), 7.41 - 7.56 (m, 3 H), 7.38 (d, 2 H), 6.41 (s, 1 H), 5.52 (br. s., 1 H), 4.28 - 4.53 (m, 1 H), 3.90 - 4.04 (m, 1 H), 3.76 - 3.89 (m, 1 H), 3.58 - 3.73 (m, 1 H), 3.41 - 3.57 (m, 1 H), 3.19 - 3.36 (m, 1 H), 2.96 - 3.17 (m, 1 H), 2.59 (s, 3 H), 1.40 (d, J=6.57 Hz, 3 H), 0.25 (s, 9 H).
2.64 Example 64 (Prepared according to Scheme 4)
[0539]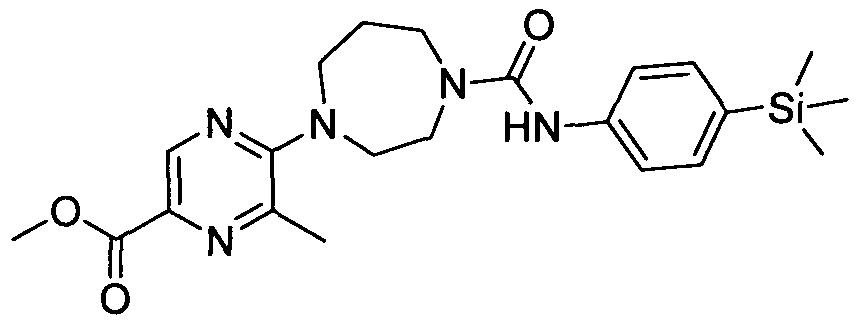 Methyl 6-methyl-5-(4-(4-(trimethylsilyl)phenylcarbamoyl)- 1 ,4-diazepan- 1 -yl)pyrazine-2-carboxylate
Methyl 6-methyl-5-(4-(4-(trimethylsilyl)phenylcarbamoyl)- 1 ,4-diazepan- 1 -yl)pyrazine-2-carboxylate
[0540]The title compound was synthesised in accordance with Example 23 using homopiperazine and methyl 5- chloro-6-methylpyrazine-2-carboxylate (CAS 77168-85-5).
[0541]MS: ES+ 442.1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1 H), 7.39 - 7.49 (m, 2 H), 7.35 (m, 2 H), 6.38 (s, 1 H),
[0542]3.97 (s, 3 H), 3.85 - 3.92 (m, 2 H), 3.74 - 3.84 (m, 4 H), 3.50 (m, 2 H), 2.64 (s, 3 H), 2.02 (m, 2 H), 0.25 (s, 9
[0544]2.65 Example 65 (Prepared according to Scheme 4)
[0545]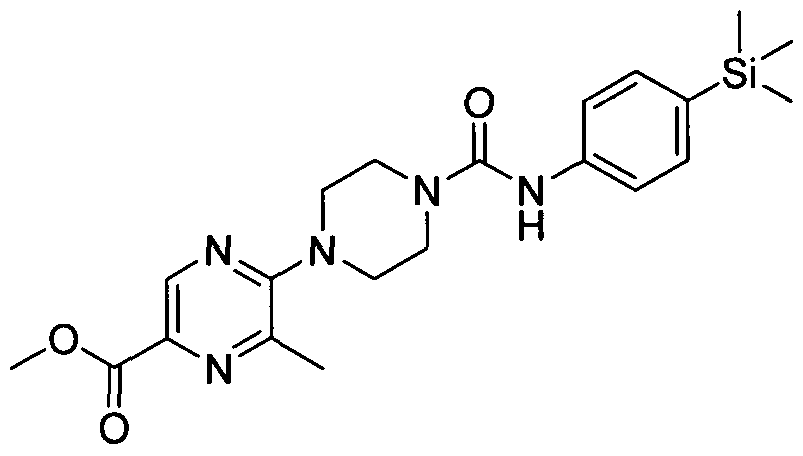
[0546]Methyl 6-methyl-5-(4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin- 1 -yl)pyrazine-2-carboxylate
[0547]The title compound was synthesised in accordance with Example 23 using piperazine and methyl 5-chloro-6- methylpyrazine-2-carboxylate (CAS 77168-85-5).
[0548]MS: ES+ 428.1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1 H), 7.41 - 7.52 (m, 2 H), 7.36 (m, 2 H), 6.41 (s, 1 H), 3.99 (s, 3 H), 3.61 - 3.78 (m, 4 H), 3.45 - 3.60 (m, 4 H), 2.63 (s, 3 H), 0.25 (s, 9 H).
[0549]2.66 Example 66 (Prepared according to Scheme 4)
[0550]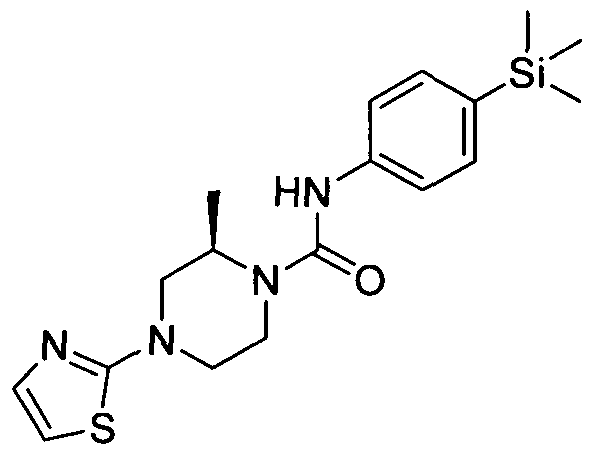
[0551](R)-2-Methyl-4-(thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
The title compound was synthesised using procedures similar to those described for Example 23 using 2- chlorothiazole.
[0552]MS: ES+ 375.1H NMR (400 MHz, DMSOd6) δ 8.60 (s, IH), 7.41 - 7.51 (m, 2H), 7.32 - 7.41 (m, 2H), 7.13
[0553]- 7.21 (m, IH), 6.82 - 6.90 (m, IH), 4.48 (br. s., IH), 3.92 - 4.03 (m, IH), 3.81 - 3.91 (m, IH), 3.67 - 3.77 (m,
[0554]IH), 3.16 - 3.28 (m, 2H), 2.97 - 3.09 (m, IH), 1.18 (s, 3H), 0.15 - 0.27 (s, 9H)
[0555]2.67 Example 67 (Prepared according to Scheme 4)
[0556]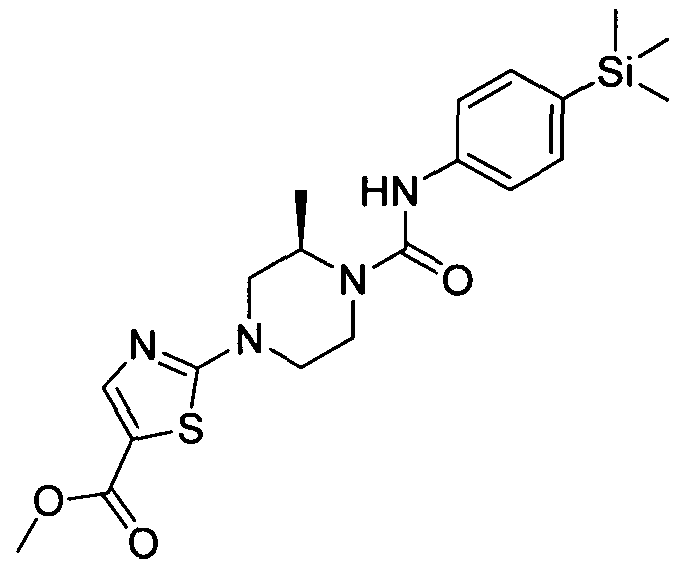
[0557](R)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylate
[0558]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine and methyl 2-chlorothiazole-5-carboxylate.
[0559]MS: ES+ 433.1H NMR (400 MHz, DMSOd6) δ 8.62 (s, IH), 7.88 (s, IH), 7.42 - 7.49 (m, 2H), 7.34 - 7.41 (m, 2H), 4.44 - 4.55 (m, IH), 3.90 - 4.07 (m, 2H), 3.78 - 3.87 (m, IH), 3.75 (s, 3H), 3.41 - 3.50 (m, IH), 3.18 - 3.31 (m, 2H), 1.09 - 1.19 (m, 3H), 0.21 (s, 9H)
[0560]2.68 Example 68 (Prepared according to Scheme 4)
[0561]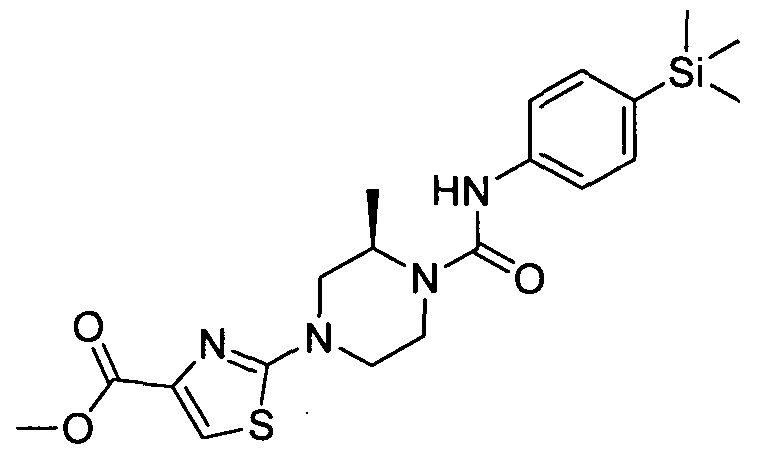
[0562](R)-Methyl2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylate
[0563]The title compound was synthesised in accordance with Example 23 using (R)-(+)-2-methylpiperazine and methyl 2-chlorothiazole-4-carboxylate.
[0564]MS: ES+ 433.1H NMR (400 MHz, DMSOd6) δ 8.62 (s, IH), 7.75 (s, IH), 7.42 - 7.49 (m, 2H), 7.34 - 7.41 (m, 2H), 4.44 - 4.54 (m, IH), 3.95 - 4.04 (m, IH), 3.83 - 3.93 (m, IH), 3.69 - 3.79 (m, 4H), 3.20 - 3.31 (m, 2H), 3.04 - 3.15 (m, IH), 1.11 - 1.22 (m, 3H), 0.21 (s, 9H)
[0565]2.69 Example 69 (Prepared according to Scheme 5, step a)
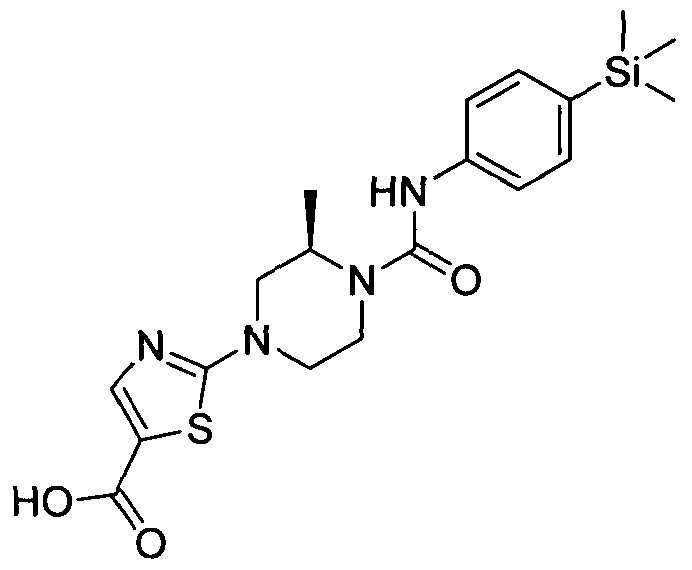 (R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylic acid
(R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylic acid
[0566](R)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylate
[0567](Example 67) was hydrolysed to the title compound using a procedure similar to that described for Example
[0569]MS: ES+ 419.1H NMR (400 MHz, DMSOd6) δ 8.61 (s, IH), 7.73 - 7.79 (m, IH), 7.42 - 7.48 (m, 2H), 7.33
[0570]- 7.40 (m, 2H), 4.49 (br. s., IH), 3.88 - 4.05 (m, 2H), 3.76 - 3.86 (m, IH), 3.38 - 3.47 (m, IH), 3.16 - 3.28 (m,
[0571]2H), 1.1 1 - 1.20 (m, 3H), 0.21 (s, 9H)
[0572]2.70 Example 70 (Prepared according Scheme 5, step a)
[0573]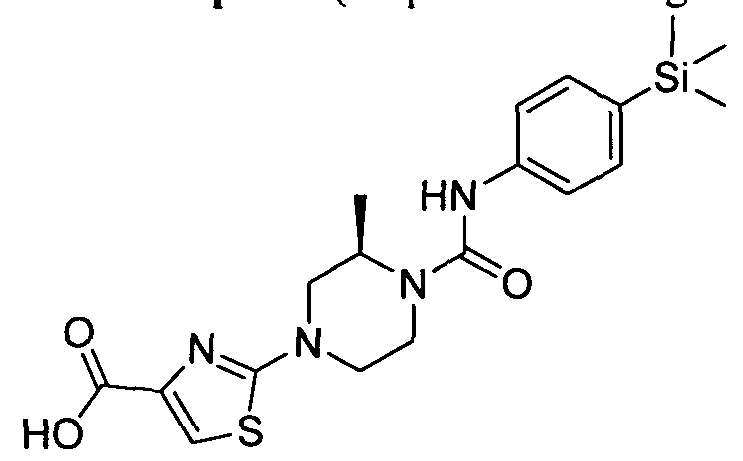 (R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylic acid
(R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylic acid
[0574](R)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylate
[0575](Example 68) was hydrolysed to the title compound using a procedure similar to that described for Example 28.
[0576]MS: ES+ 419.1H NMR (400 MHz, DMSOd6) δ 8.61 (s, IH), 7.65 (s, IH), 7.43 - 7.49 (m, 2H), 7.33 - 7.41 (m, 2H), 4.49 (br. s., IH), 3.94 - 4.04 (m, IH), 3.84 - 3.93 (m, IH), 3.68 - 3.76 (m, IH), 3.18 - 3.29 (m, 2H), 3.02 - 3.14 (m, IH), 1.14 - 1.22 (m, 3H), 0.18 (s, 9H)
[0577]2.71 Example 71 (Prepared according to Scheme 6, step a)
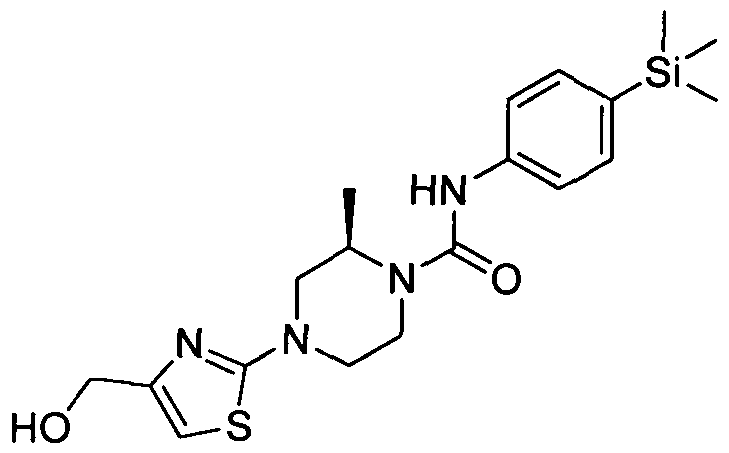 (R)-4-(4-(Hydroxymethyl)thiazol-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
(R)-4-(4-(Hydroxymethyl)thiazol-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0578]The title compound was synthesised using a procedure similar to that described for Example 31 starting from (R)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin- 1 -yl)thiazole-4-carboxylate (Example 68).
[0579]MS: ES+ 405.1H NMR (400 MHz, DMSO-J6) δ ppm 8.58 (s, 1 H) 7.42 - 7.52 (m, 2 H) 7.34 - 7.42 (m, 2 H) 6.56 (s, 1 H) 5.05 - 5.16 (m, 1 H) 4.48 (br. s., 1 H) 4.30 - 4.40 (m, 2 H) 4.00 (br. s., 1 H) 3.85 (br. s., 1 H) 3.61 - 3.74 (m, 1 H) 3.17 - 3.28 (m, 2 H) 2.94 - 3.08 (m, 1 H) 1.13 - 1.26 (m, 3 H) 0.21 (s, 9 H)
[0580]2.72 Example 72 (Prepared according to Scheme 5, step b)
[0581]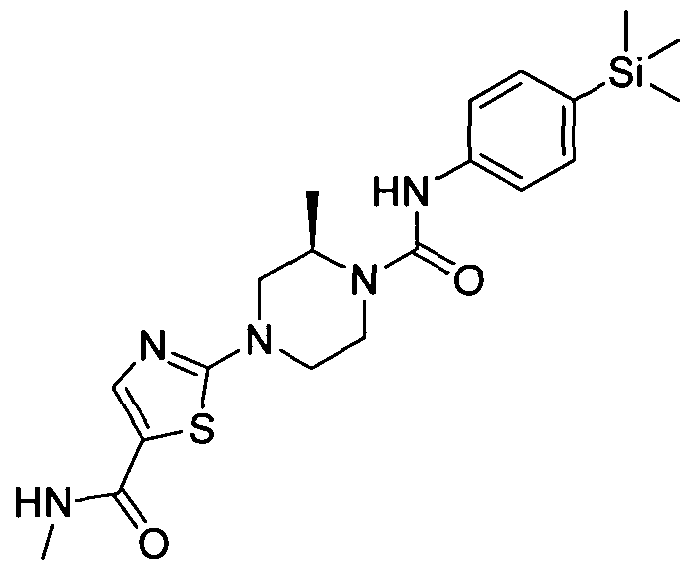 (R)-N-Methyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxamide
(R)-N-Methyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxamide
[0582]The title compound was synthesised using a procedure similar to that described for Example 30 starting from
[0583](R)-2-(3-Methy l-4-(4-(trimethylsilyl)phenylcarbarnoyl)piperazin- 1 -yI)thiazole-5-carboxy!ic acid (Example
[0585]MS: ES+ 432.1H NMR (400 MHz, DMSO-J6) δ ppm 8.57 (br. s., 1 H) 8.10 - 8.20 (m, 1 H) 7.74 (s, 1 H)
[0586]7.42 - 7.50 (m, 2 H) 7.33 - 7.41 (m, 2 H) 4.48 (br. s., 1 H) 3.94 - 4.04 (m, 1 H) 3.86 - 3.95 (m, 1 H) 3.72 -
[0587]3.82 (m, 1 H) 3.33 - 3.42 (m, 1 H) 3.08 - 3.26 (m, 2 H) 2.67 - 2.75 (m, 3 H) 1.12 - 1.19 (m, 3 H) 0.21 (s, 9 H)
[0588]2.73 Example 73 (Prepared according to Scheme 5, step b)
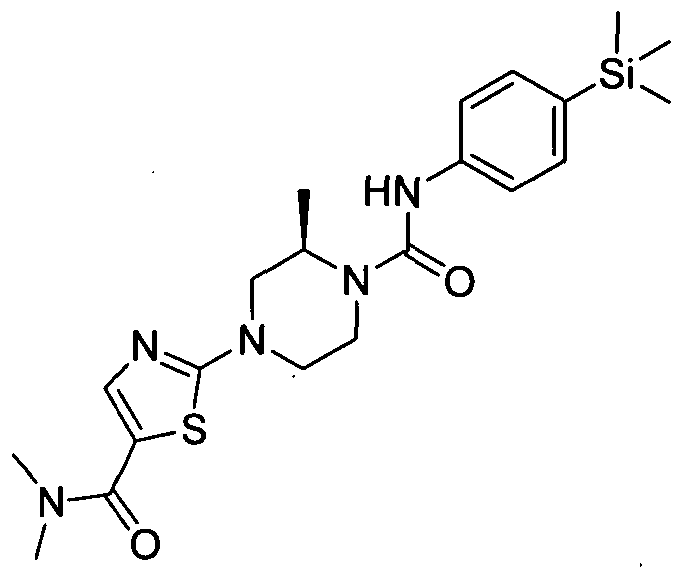
[0589](R)-N,N-Dimethyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxamide
[0590]The title compound was synthesised using a procedure similar to that described for Example 30 starting from
[0591](R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylic acid (Example
[0593]MS: ES+ 446.1H NMR (400 MHz, DMSO-J6) δ ppm 8.58 (s, 1 H) 7.60 (s, 1 H) 7.41 - 7.49 (m, 2 H) 7.33 -
[0594]7.41 (m, 2 H) 4.49 (br. s., 1 H) 3.95 - 4.03 (m, 1 H) 3.87 - 3.95 (m, 1 H) 3.74 - 3.82 (m, 1 H) 3.33 - 3.41 (m, 1
[0595]H) 3.13 - 3.26 (m, 2 H) 3.08 (br. s., 6 H) 1.11 - 1.20 (m, 3 H) 0.21 (s, 9 H)
[0596]2.74 Example 74 (Prepared according to Scheme 5, step b)
[0597]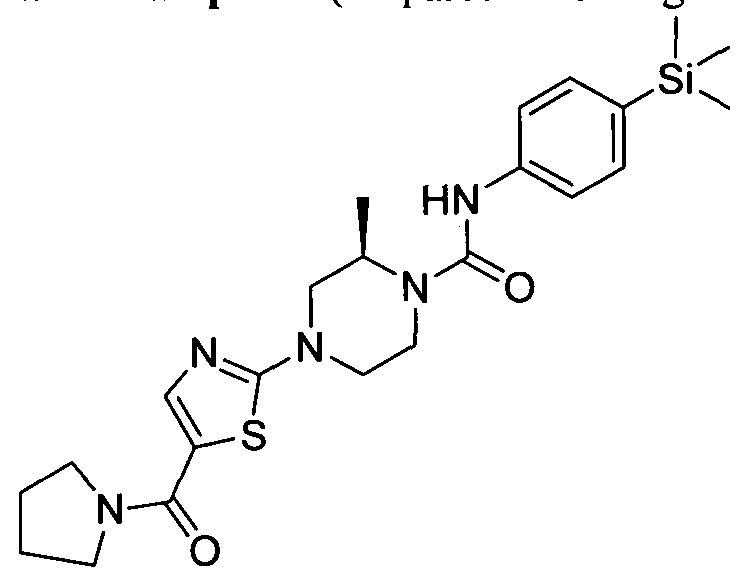
[0598](R)-2-Methyl-4-(5-(pyrrolidine-l-carbonyl)thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0599]The title compound was synthesised using a procedure similar to that described for Example 30 starting from
[0600](R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylic acid (Example
[0602]MS: ES+ 472.1H NMR (400 MHz, DMSO-J6) δ ppm 8.57 (br. s., 1 H) 7.64 (s, 1 H) 7.41 - 7.50 (m, 2 H)
[0603]7.32 - 7.41 (m, 2 H) 4.43 - 4.55 (m, 1 H) 3.88 - 4.04 (m, 2 H) 3.74 - 3.84 (m, 1 H) 3.64 (br. s., 2 H) 3.33 -
[0604]3.52 (m, 3 H) 3.10 - 3.26 (m, 2 H) 1.73 - 2.03 (m, 4 H) 1.11 - 1.20 (m, 3 H) 0.21 (s, 9 H)
[0605]2.75 Example 75 (Prepared according to Scheme 5, step b)
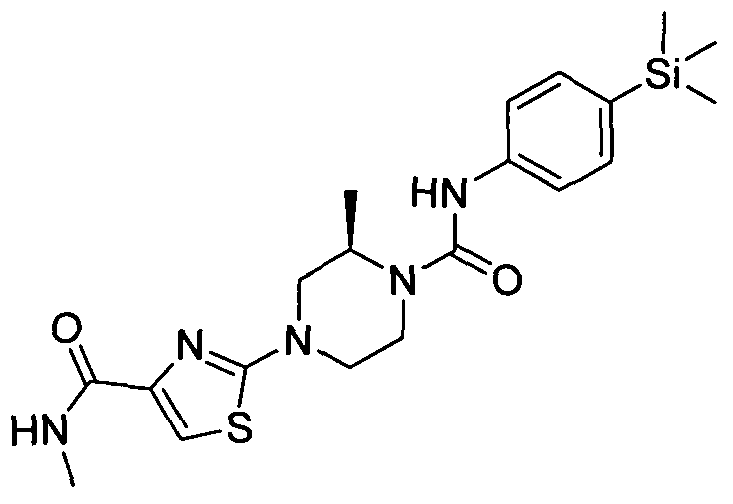 (R)-N-Methyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxamide
(R)-N-Methyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxamide
[0606]The title compound was synthesised using a procedure similar to that described for Example 30 starting from(R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylic acid (Example 70).
[0607]MS: ES+ 432.1H NMR (400 MHz, DMSCMO δ ppm 8.62 (s, 1 H) 8.03 - 8.12 (m, 1 H) 7.42 - 7.50 (m, 2 H) 7.33 - 7.41 (m, 3 H) 4.49 (br. s., 1 H) 3.90 - 4.05 (m, 2 H) 3.74 - 3.84 (m, 1 H) 3.19 - 3.31 (m, 2 H) 3.02 - 3.15 (m, 1 H) 2.70 - 2.79 (m, 3 H) 1.14 - 1.23 (m, 3 H) 0.21 (s, 9 H)
[0608]2.76 Example 76 (Prepared according to Scheme step b)
[0609]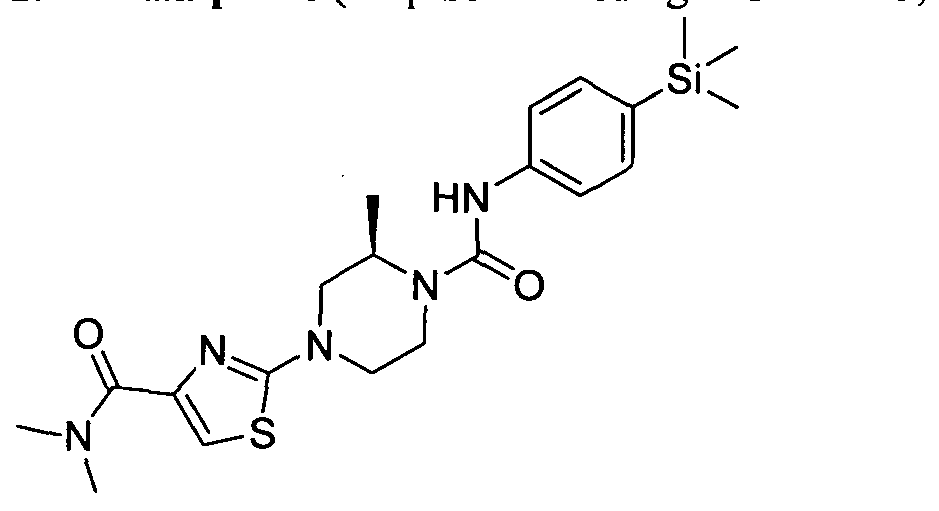 (R)-N,N-Dimethyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxamide
(R)-N,N-Dimethyl-2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxamide
[0610]The title compound was synthesised using a procedure similar to that described for Example 30 starting from
[0611](R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-4-carboxylic acid (Example
[0613]MS: ES+ 446.1H NMR (400 MHz, DMSO-c4) δ ppm 8.61 (s, 1 H) 7.42 - 7.49 (m, 2 H) 7.34 - 7.41 (m, 2 H)
[0614]7.19 (s, 1 H) 4.44 - 4.53 (m, 1 H) 3.94 - 4.03 (m, 1 H) 3.81 - 3.89 (m, 1 H) 3.68 - 3.77 (m, 1 H) 3.19 - 3.32 (m,
[0615]2 H) 3.03 - 3.15 (m, 4 H) 2.93 (br. s., 3 H) 1.14 - 1.21 (m, 3 H) 0.21 (s, 9 H)
[0616]2.77 Example 77 (Prepared according to 5, step b)
[0617]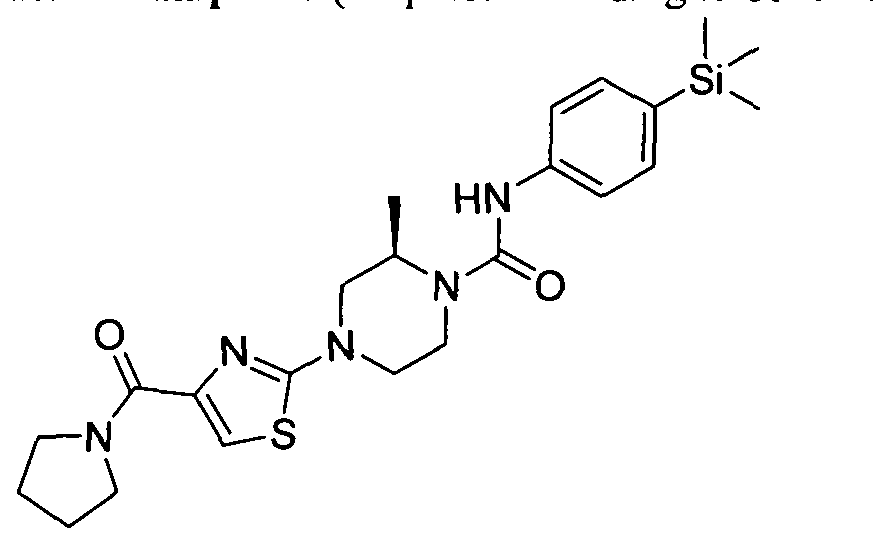 (R)-2-Methyl-4-(4-(pyrrolidine- 1 -carbonyl)thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 - carboxamide
(R)-2-Methyl-4-(4-(pyrrolidine- 1 -carbonyl)thiazol-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine- 1 - carboxamide
[0618]The title compound was synthesised using a procedure similar to that described for Example 30 starting from
[0619](R)-2-(3-Methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin- 1 -yl)thiazole-4-carboxylic acid (Example
[0621]MS: ES+ 472.1H NMR (400 MHz, DMSCW6) δ ppm 8.58 (s, 1 H) 7.42 - 7.49 (m, 2 H) 7.34 - 7.41 (m, 2 H)
[0622]7.32 (s, 1 H) 4.49 (br. s., 1 H) 3.94 - 4.04 (m, 1 H) 3.82 - 3.91 (m, 1 H) 3.66 - 3.79 (m, 3 H) 3.39 - 3.48 (m, 2
[0623]H) 3.23 (br. s., 2 H) 3.03 - 3.15 (m, 1 H) 1.73 - 1.91 (m, 4 H) 1.13 - 1.22 (m, 3 H) 0.21 (s, 9 H)
[0624]2.78 Example 78 (Prepared according to Scheme 6, step a)
[0625]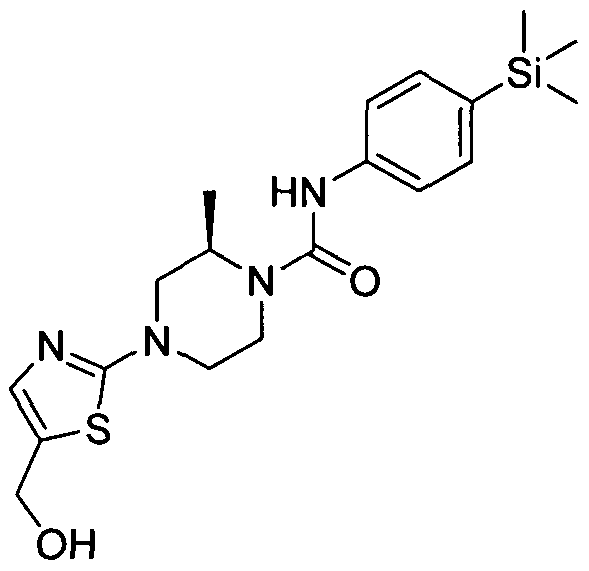
[0626](R)-4-(5-(Hydroxymethyl)thiazol-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0627]The title compound was synthesised using a procedure similar to that described for Example 31 starting from (R)-Methyl 2-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)thiazole-5-carboxylate (Example 67).
[0628]MS: ES+ 405.1H NMR (400 MHz, DMSO-^5) δ ppm 8.58 (s, 1 H) 7.42 - 7.49 (m, 2 H) 7.33 - 7.41 (m, 2 H) 6.99 (s, 1 H) 5.18 - 5.26 (m, 1 H) 4.41 - 4.52 (m, 3 H) 3.92 - 4.03 (m, 1 H) 3.78 - 3.89 (m, 1 H) 3.64 - 3.74 (m, 1 H) 3.15 - 3.27 (m, 2 H) 2.96 - 3.08 (m, 1 H) 1.11 - 1.22 (m, 3 H) 0.21 (s, 9 H)
[0629]2.79 Example 79 (Prepared according to Scheme 4)
[0630]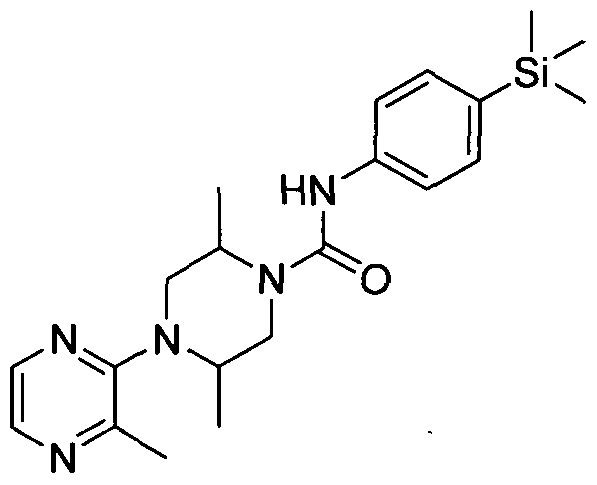
[0631]2,5-Dimethyl-4-(3-methylpyrazin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l -carboxamide
[0632]The title compound was synthesised using a procedure similar to that described for Example 23 using 2- chloro-3-methylpyrazine and (2S,5R)-2,5-dimethylpiperazine.
MS: ES+ 398.1H NMR (400 MHz, DMSCMs) δ ppm 8.48 (s, 1 H) 8.06 - 8.1 1 (m, 1 H) 8.01 - 8.06 (m, 1 H) 7.44 - 7.52 (m, 2 H) 7.32 - 7.41 (m, 2 H) 4.41 - 4.52 (m, 1 H) 3.96 - 4.07 (m, 1 H) 3.76 - 3.85 (m, 1 H) 3.44 - 3.58 (m, 2 H) 3.24 - 3.35 (m, 4 H) 3.08 - 3.17 (m, 1 H) 1.15 - 1.24 (m, 3 H) 0.95 - 1.02 (m, 3 H) 0.20 (s, 9 H)
[0633]2.80 Example 80 (Prepared according to Scheme 4)
[0634]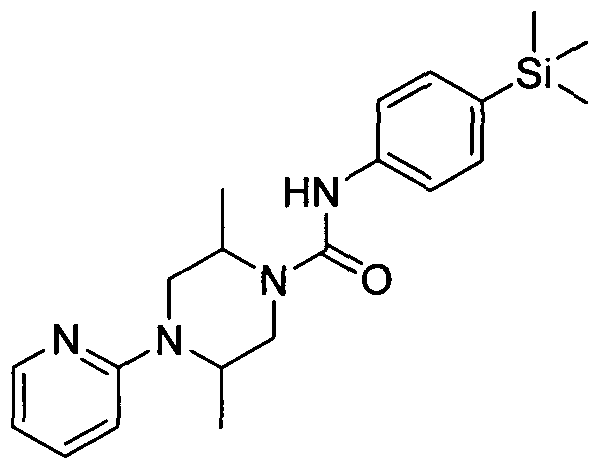
[0635]2,5-Dimethyl-4-(pyridin-2-yl)-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0636]The title compound was synthesised using a procedure similar to that described for Example 23 using 2- chloropyridine and 2,5-dimethylpiperazine.
[0637]MS: ES+ 398.1H NMR (400 MHz, DMSCW6) δ ppm 8.26 - 8.36 (m, 1 H) 7.85 - 7.93 (m, 1 H) 7.22 - 7.36 (m, 3 H) 7.10 - 7.21 (m, 2 H) 6.56 - 6.66 (m, 1 H) 6.34 - 6.44 (m, 1 H) 4.20 - 4.40 (m, 2 H) 3.58 - 3.81 (m, 2 H) 3.11 - 3.18 (m, 1 H) 2.97 - 3.06 (m, 1 H) 0.89 - 0.98 (m, 3 H) 0.79 - 0.89 (m, 3 H) -0.06 - 0.05 (m, 9 H).
[0638]2.81 Example 81 (Prepared according to Scheme 4)
[0639]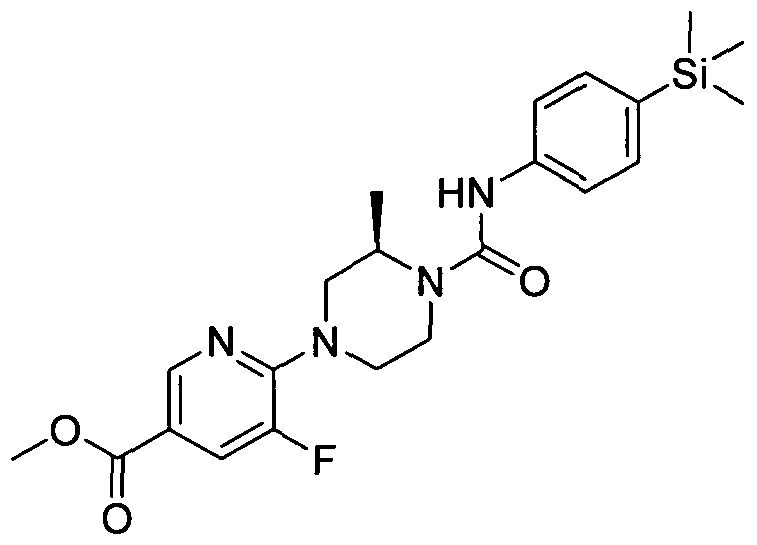 (R)-Methyl-5-fluoro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate
(R)-Methyl-5-fluoro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate
[0640]The title compound was synthesised using a procedure similar to that described for Example 23 starting from
[0641](R)-(+)-2-methylpiperazine.
[0642]MS: ES+ 445.1H NMR (400 MHz, DMSO-J6) δ ppm 8.49 (s, 1 H) 8.32 - 8.40 (m, 1 H) 7.86 - 7.93 (m, 1 H)
[0643]7.42 - 7.51 (m, 2 H) 7.32 - 7.40 (m, 2 H) 4.37 - 4.46 (m, 1 H) 3.80 - 3.93 (m, 4 H) 3.50 - 3.60 (m, 2 H) 3.25 -
[0644]3.36 (m, 1 H) 3.13 - 3.23 (m, 1 H) 2.86 - 2.97 (m, 1 H) 1.13 - 1.21 (m, 3 H) 0.16 - 0.25 (m, 9 H)
[0645]2.82 Example 82 (Prepared according to Scheme 4)
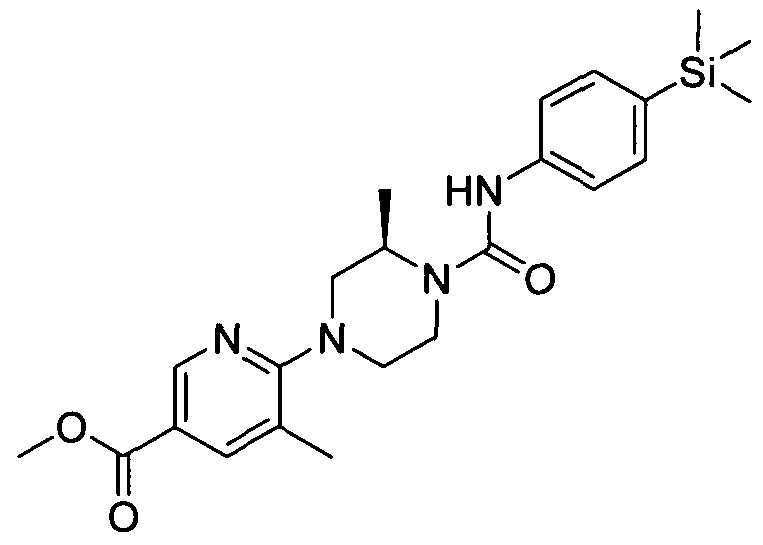
[0646](R)-Methyl-5-methyl-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinate
[0647]The title compound was synthesised using a procedure similar to that described for Example 23 starting from
[0648](i?)-(+)-2-methylpiperazine.
[0649]MS: ES+ 441.1H NMR (400 MHz, OMSO-d6) δ ppm 8.60 - 8.68 (m, 1 H) 8.54 (s, 1 H) 7.92 - 7.99 (m, 1 H)
[0650]7.43 - 7.51 (m, 2 H) 7.33 - 7.41 (m, 2 H) 4.40 - 4.52 (m, 1 H) 3.92 - 4.03 (m, 1 H) 3.83 (s, 3 H) 3.68 - 3.77 (m,
[0651]1 H) 3.56 - 3.65 (m, 1 H) 3.23 - 3.28 (m, 1 H) 2.98 - 3.09 (m, 1 H) 2.82 - 2.95 (m, 1 H) 2.35 (s, 3 H) 1.19 -
[0652]1.30 (m, 3 H) 0.21 (s, 9 H)
[0653]2.83 Example 83 (Prepared according to Scheme 11, steps c,d,e, f and g)
[0654]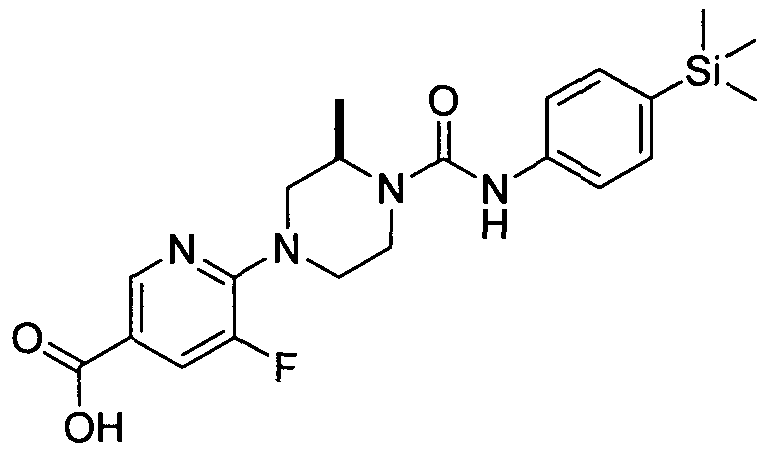 (R)-5-Fluoro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
(R)-5-Fluoro-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
[0655]Synthesized in accordance with procedures outlined in Example 51, from ό-chloro-S-fluoronicotinonitrile. MS: ES+ 431.2.1H NMR (400 MHz, CDCl3) δ 8.70 - 9.10 (m, IH), 8.06 (s, IH), 7.43 - 7.51 (m, 2H), 7.36 - 7.43 (m, 2H), 6.44 - 6.65 (m, IH), 4.45 (br. s., IH), 3.93 - 4.06 (m, IH), 3.74 - 3.90 (m, I H), 3.62 - 3.74 (m, IH), 3.45 - 3.59 (m, IH), 3.36 (br. s., IH), 3.15 (br. s., IH), 2.41 (s, 3H), 1.36 - 1.48 (m, 3H), 0.27 (s, 9H)
[0656]2.84 Example 84 (Prepared according to Scheme 11, steps c, d, e and f)
[0657]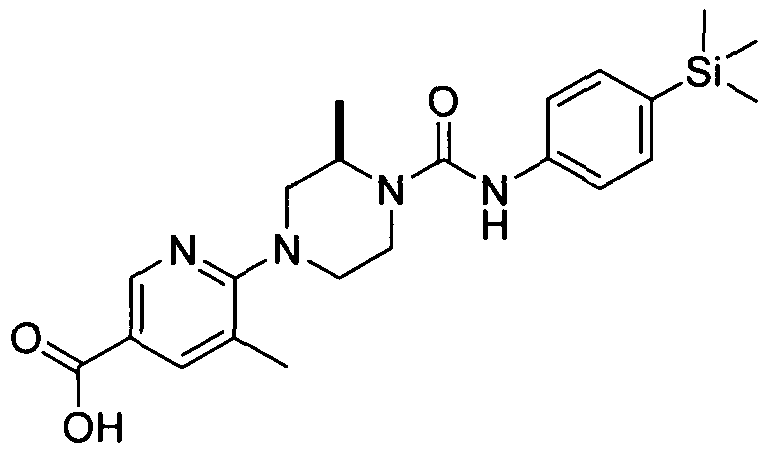 (R)-5-methyl-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
Synthesized in accordance with procedures outlined in Example 51 from, commercially available, 6-chloro-
(R)-5-methyl-6-(3-methyl-4-(4-(trimethylsilyl)phenylcarbamoyl)piperazin-l-yl)nicotinic acid
Synthesized in accordance with procedures outlined in Example 51 from, commercially available, 6-chloro-
[0658]5-methylnicotionitrile.
[0659]MS: ES+ 427.2.1H NfMR (400 MHz, CDCl3) δ 8.76 - 8.99 (m, IH), 8.00 - 8.17 (m, IH), 7.43 - 7.53 (m, 2H),
[0660]7.37 - 7.43 (m, 2H), 6.50 (br. s., IH), 4.44 (br. s., IH), 3.93 - 4.03 (m, IH), 3.79 - 3.89 (m, IH), 3.60 - 3.71
[0661](m, IH), 3.44 - 3.59 (m, IH), 3.27 - 3.39 (m, IH), 3.04 - 3.22 (m, IH), 2.40 (s, 3H), 1.38 - 1.47 (m, 3H), 0.27
[0663]2.85 Example 85 (Prepared according to Scheme 15)
[0664]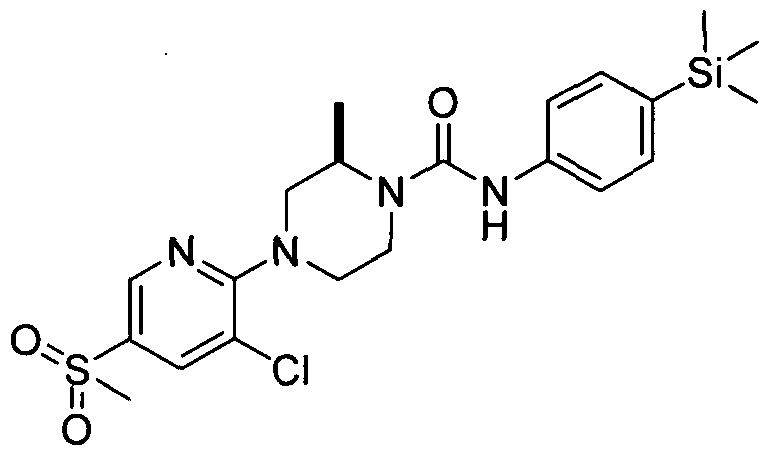
[0665](/?)-4-(3-Chloro-5-(methylsulfonyl)pyridine-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l- carboxamide
[0666]To a solution of 3-amino-5,6-dichloropyridine (6.13mmol) in HCl (37%, 5ml) at 00C was added sodium nitrite (9.20mmol) in water (2ml) in a dropwise fashion. The reaction was stirred at 00C for 1 hr. The reaction was filtered to remove sodium chloride. A suspension of sodium methanethiolate (7.36mmol) and copper (II) tetrafluoroborate hydrate (0.061mmol) in acetonitrile (5 ml) was cooled to 00C. The solution of the diazonium salt was added dropwise with much nitrogen evolution. The resulting orange suspension was stirred at room temperature for 4 hours. The reaction was quenched with NaOH (50ml) and extracted with ethyl acetate (2x50ml). The organics were combined, washed with brine, dried (MgSO4) and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (0-30% ethyl acetate in petroleum ether) yielding the product 2,3-dichloro-5-(methylthio)pyridine (3.77mmol) 1H ΝMR (400 MHz, CDCl3) δ 8.16 - 8.34 (m, IH), 7.59 - 7.76 (m, IH), 2.59 (s, 3H)
[0667]A solution of 2,3-dichloro-5-(methylthio)pyridine (3.77mmol) in DCM was cooled to 00C. To this was added mCPBA (7.53mmol) the reaction was warmed to room temperature and stirred for 48hr. The reaction was quenched with sat. sodium bicarbonate (50 ml) and extracted with DCM (50 ml x 2) the organic phases were combined, washed with brine, dried (phase separation cartridge) and concentrated under reduced pressure. The resultant white solid was purified by flash chromatography (0-30 % ethyl acetate in petroleum ether) yielding 2,3-dichloro-5-(methylsulfonyl)pyridine (3.72mmol). 1H ΝMR (400 MHz, CDCl3) δ 8.61 - 8.99 (m, IH), 8.09 - 8.47 (m, IH), 3.08 (s, 3H).
[0668]The title compound was synthesized in accordance with Example 23 from the intermediate described above. MS: ES+ 481.1.1H ΝMR (400 MHz, CDCl3) δ 8.62 - 8.69 (m, IH), 8.03 - 8.10 (m, IH), 7.43 - 7.55 (m, 2H), 7.32 - 7.43 (m, 2H), 6.37 (s, IH), 4.35 - 4.48 (m, IH), 4.17 - 4.29 (m, IH), 4.05 - 4.17 (m, IH), 3.93 - 4.03 (m,
IH), 3.43 - 3.57 (m, IH), 3.33 - 3.43 (m, IH), 3.13 - 3.26 (m, IH), 3.1 1 (s, 3H), 1.35 - 1.46 (m, 3H), 0.27 (s, 9H)
[0669]2.86 Example 86 (Prepared according to Scheme 16)
[0670]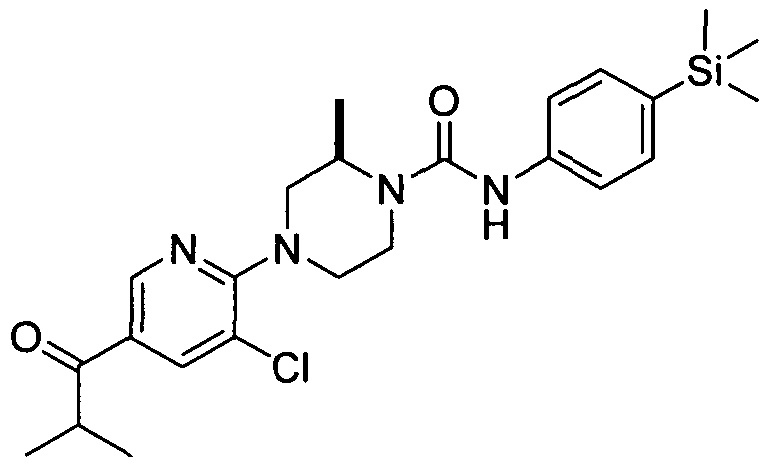
[0671](R)-4-(3-Chloro-5-isobutyrylpyridin-2-yl)-2-methyl-N-(4-(trimethylsilyl)phenyl)piperazine-l-carboxamide
[0672]To a solution of 5,6-dichloronicotinoyl chloride (4.75mmol) in THF (50 ml) at -780C was added isopropylmagnesium chloride (6.18mmol) in a dropwise fashion. The reaction was stirred at -78 °C for 1 hour. The reaction was quenched with saturated ammonium chloride (15ml) followed by water (15ml) and extracted with ethyl acetate (30 ml). The organic was separated, dried (phase separation cartridge) and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (0-20 % ethyl acetate in petroleum ether yielding the product l-(5,6-dichloropyridin-3-yl)-2-methylpropan-l-one
[0674]1H ΝMR (400 MHz, DMSOd6) δ 8.82 - 9.01 (m, IH), 8.52 - 8.65 (m, I H), 3.54 - 3.82 (m, IH), 1.04 - 1.19
[0676]The title compound was synthesized in accordance with Example 23 from l-(5,6-dichloropyridin-3-yl)-2- methy lpropan- 1 -one.
[0677]MS: ES+ 473.3.1H ΝMR (400 MHz, CDCl3) δ 8.58 - 8.66 (m, IH), 8.02 - 8.10 (m, IH), 7.31 - 7.42 (m, 2H),
[0678]7.23 - 7.31 (m, 2H), 6.26 (s, IH), 4.23 - 4.35 (m, IH), 3.92 - 4.12 (m, 2H), 3.78 - 3.90 (m, IH), 3.26 - 3.47 (m,
[0679]2H), 3.13 - 3.25 (m, IH), 2.94 - 3.09 (m, IH), 1.27 - 1.35 (m, 3H), 1.04 - 1.18 (m, 6H), 0.15 (s, 9H)
[0680]3. Biological efficacy of compounds of the invention 3.1 In vitro TrpVl binding assay
[0681]A number of TrpVl antagonist compounds of the invention were functionally assessed by measurement of the change in intracellular calcium levels induced by the influx of extracellular calcium in response to the TrpVl activator, capsaicin. The ability of compounds to block the influx of calcium by capsaicin in CHO-Kl cells expressing the human TrpVl channel were determined as a measure of the compound's antagonist activity in vitro.
[0682]CHO-Kl cells (20,000), expressing the human TrpVl channel, were seeded in 30μl of normal culture medium into each well of a 384 well assay plate (Black, clear bottom - Greiner) and incubated overnight at standard culture conditions (370C, 5% CO2). Following the overnight incubation the media was aspirated and
replaced with 45μl assay buffer (IX Hanks buffered saline, 25mM HEPES, 0.1% w/v fatty acid free BSA (bovine serum albumen), pH7.4) containing 2.5mM probenecid, 0.08% pluronic acid and 4μM Fluo-4. Cells were incubated at 370C for 1 hour 10 minutes to allow for dye uptake after which the dye containing buffer was replaced with an equal volume of assay buffer alone (IX Hanks buffered saline, 25mM HEPES, 0.1% w/v fatty acid free BSA).
[0683]To test for antagonist activity, compounds at a final concentration range between 0. InM - 3.2μM (diluted in assay buffer) were added to the assay wells in a volume of 5μl and allowed to incubate 15 minutes prior to stimulation with capsaicin. After incubation with test compounds the assay plate was placed in a FLIPR Tetra (Molecular Devices) and capsaicin (diluted in assay buffer) was added at the determined EC50 concentration (1OnM final). Capsaicin-dependent changes in intracellular calcium levels were determined by measuring changes in fluorescence of the dye between 515-575nM following excitation between 470-495nM.
[0684]Readings from wells that do not contain antagonist allow percentage inhibition curves to be plotted using 4- parameter fit algorithm and the calculation of IC50 values for each compound.
[0686]The results of the biological assay carried out in 3.1 were as follows:
[0687]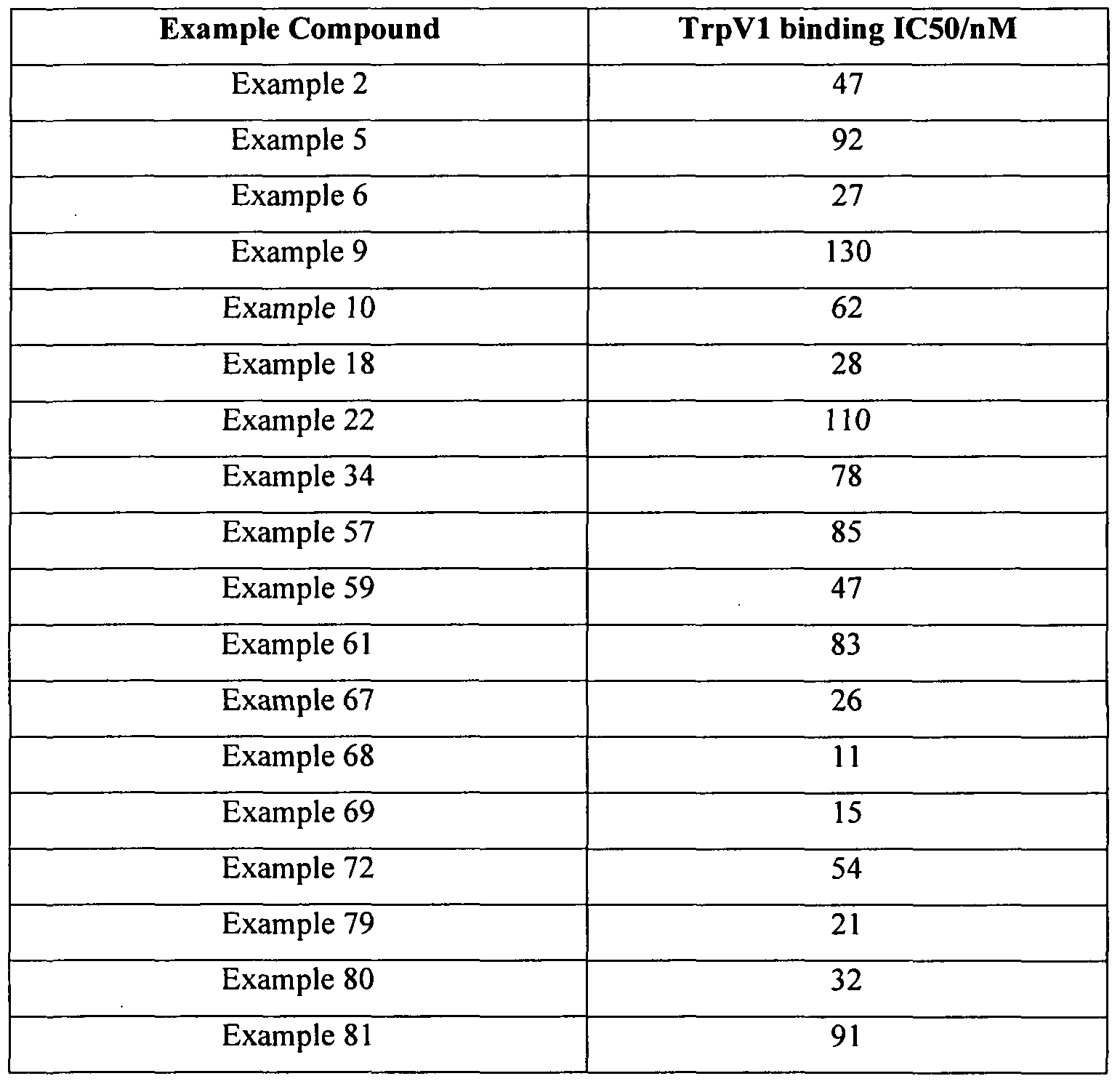 These results indicate that compounds of the invention have potent antagonist activity at the TrpVl receptor. The compounds tested exhibit IC50 values of less than 10 μM, in many cases less than 4μM, and several compounds show affinity at the TpVl receptor in the low nanomolar range. Accordingly the compounds of the invention are expected to have usefulness in the prevention or treatment of conditions, such as those discussed above, in which the TrpVl receptor is implicated.
These results indicate that compounds of the invention have potent antagonist activity at the TrpVl receptor. The compounds tested exhibit IC50 values of less than 10 μM, in many cases less than 4μM, and several compounds show affinity at the TpVl receptor in the low nanomolar range. Accordingly the compounds of the invention are expected to have usefulness in the prevention or treatment of conditions, such as those discussed above, in which the TrpVl receptor is implicated.
[0689]1. Caterina MJ. et al, Nature, 1997, 389, 816-824
[0690]2. Barton N.J. et al, Exp. MoI. Pathol., 2006, 81, 166-170
[0691]3. Catarina M.J. et al, Science, 2000, 288, 5464, 306-313
[0692]4. Davis J.B. et al, Nature, 2000, 405, 183-187
[0693]5. Rami H.K. et al 2006, Bioorg. Med. Chem. Lett., 16, 3287-3291
[0694]6. Park CK et al, 2006, J. Biol. Chem., 281, 17304-17311.
[0695]7. Geppetti P et al, 2004, Br. J. Pharmacol., 141(8), 1313-1320
[0696]8. Liddle R.A. et al 2004, Pancreatomy, 4(6), 551-560
[0697]9. Nilius B et al, Physiol. Rev., 2007, 87, 165-217
[0698]10. Karlsson JA et al, 1993, Thorax, 48(4), 396-400
[0699]1 1. Jia Y et al 2002, Br. J. Pharmacol., 137, 831-836
[0700]12. O'Connell F et al 1996, Respir. Med. 90 279-286; Nakajima T et al, 2006, Allergol. Int., 55, 149-155
[0701]13. Weinfeld D et al, 2002, Ann. Allergy Asthma Immunol. 89, 419-424.
[0702]14. Birder LA et al, Nat. Neurosci., 2002, 5, 856-860
[0703]15. Charrua A et al J. Urol. 2009, 181, 379-386
[0704]16. Razavi et al 2006, Cell, 127, 1123-1135
[0705]17. Gram D.X., et al 2007, Eur. J. Neurosci. 25, 231-223
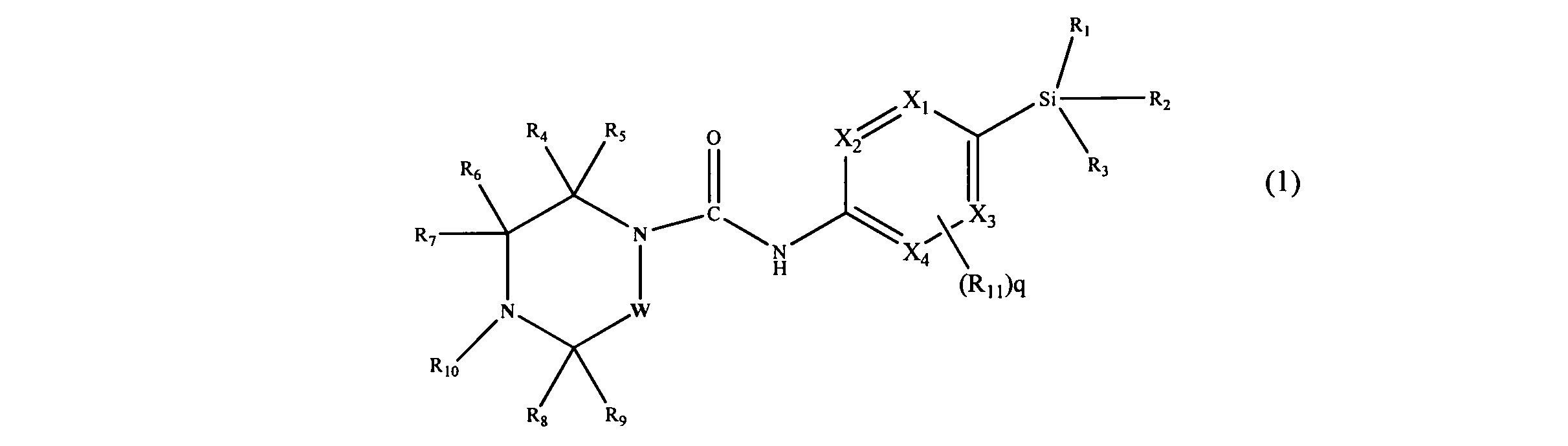
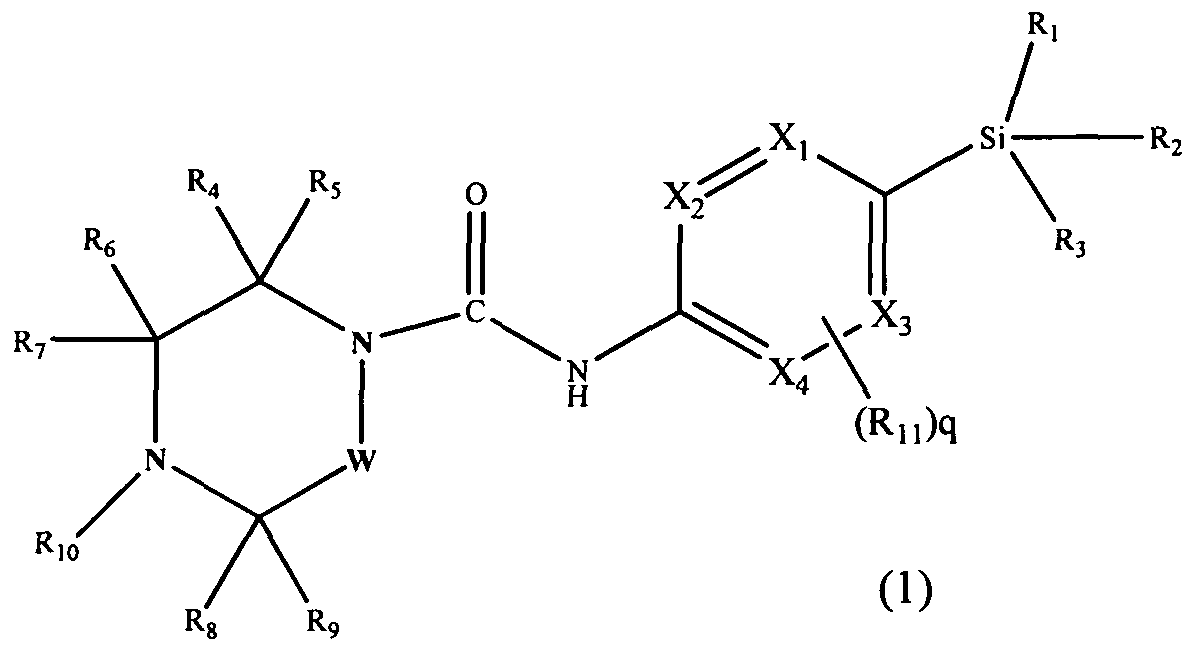 wherein:
wherein: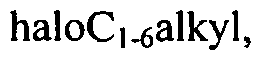 C)-6alkoxy,
C)-6alkoxy,
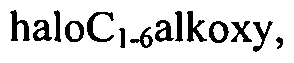 cyano or Ci-6 alkyl which may be
optionally substituted with one or more substituents independently selected from halogen, haloCi-6 alkyl and OR16; q represents 0, 1 or 2;
cyano or Ci-6 alkyl which may be
optionally substituted with one or more substituents independently selected from halogen, haloCi-6 alkyl and OR16; q represents 0, 1 or 2;
